Actualizaciones de Asuntos Regulatorios
Descubre las últimas noticias en Asuntos Regulatorios
Artículo 5.5 Aplicación del IVDR a los productos internos
- Kirsten Van Garsse, Authorized Representative Director & Regulatory Affairs Manager

La clave para entender el Artículo 5(5) es que tales pruebas sólo pueden ser realizadas por las denominadas instituciones sanitarias, que son instalaciones establecidas en la Unión Europea en las que el propósito principal es el cuidado o tratamiento de pacientes o la promoción de la salud pública(MDCG 2023-01) SI no hay disponible un dispositivo comercialmente equivalente.
Para una visión general del calendario de aplicación de las disposiciones del apartado 5 del artículo 5, consulta la Figura 1.
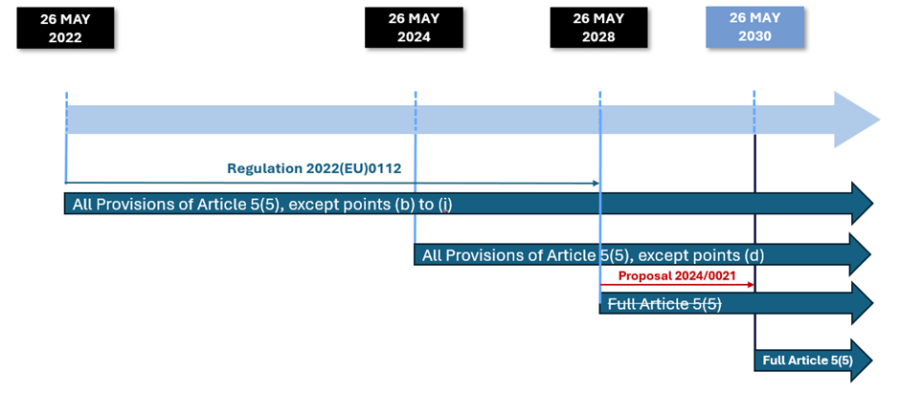
Figura 1: Calendario de aplicación de las distintas disposiciones del artículo 5(5) del DIVR
Requisitos que ya se aplicaban a todos los DIV internos desde el 26 de mayo de 2022:
- los requisitos generales de seguridad y rendimiento (RGSR) establecidos en el Anexo I;
- los dispositivos no se transfieran a otra entidad jurídica;
Requisitos que entrarán en vigor a partir de ahora (26 de mayo de 2024):
- La fabricación y el uso de los productos se realizan con arreglo a sistemas de gestión de la calidad adecuados;
- el laboratorio de la institución sanitaria (IH) cumple la norma EN ISO 15189 o, en su caso, las disposiciones nacionales en materia de acreditación
- la institución sanitaria facilite a su autoridad competente, previa solicitud, información sobre la utilización de dichos productos, que incluirá una justificación de su fabricación, modificación y utilización;
- la institución sanitaria elabora una declaración que pondrá a disposición del público, en la que se incluirá:
-
- el nombre y la dirección de la institución sanitaria fabricante,
- los detalles necesarios para identificar los dispositivos,
- una declaración de que los productos cumplen los requisitos generales de seguridad y funcionamiento establecidos en el anexo I del presente Reglamento y, en su caso, información sobre qué requisitos no se cumplen plenamente, con una justificación razonada;
- por lo que respecta a los productos de la clase D, de conformidad con las normas establecidas en el anexo VIII, la institución sanitaria elabore una documentación que permita conocer las instalaciones de fabricación, el proceso de fabricación, los datos sobre el diseño y las prestaciones de los productos, incluida la finalidad prevista, y que sea lo suficientemente detallada como para permitir a la autoridad competente cerciorarse de que se cumplen los requisitos generales de seguridad y prestaciones establecidos en el anexo I del presente Reglamento.
Los Estados miembros podrán aplicar esta disposición también a los productos de las clases A, B o C, de conformidad con las normas establecidas en el anexo VIII;
- la institución sanitaria adopte todas las medidas necesarias para garantizar que todos los productos se fabriquen de conformidad con la documentación antes mencionada y
- la institución sanitaria revisa la experiencia adquirida con el uso clínico de los productos y adopta todas las medidas correctoras necesarias;
Lo ideal es que los fabricantes dispongan de un procedimiento documentado para recopilar datos clínicos y de rendimiento , y para procesar los incidentes y las medidas correctoras del DHI.
Requisitos que se aplicarán a partir del 26 de mayo de 2028 (se ampliarán hasta 2030 según la última propuesta de reglamento de modificación*)
- la institución sanitaria justifica en su documentación que las necesidades específicas del grupo de pacientes destinatarios no pueden satisfacerse, o no pueden satisfacerse al nivel adecuado de prestaciones, mediante un dispositivo equivalente disponible en el mercado;
*Este requisito no se aplicará hasta el 26 de mayo de 2030, según la última propuesta 2024/0021 de ampliar las disposiciones transitorias del IVDR para garantizar que todos los fabricantes hayan completado su transición al IVDR.
Requiere una comparación del rendimiento con las afirmaciones del IVDR en las Instrucciones de Uso (IFU), pero como no todos los fabricantes han completado su transición, esto no puede hacerse todavía.
¿Qué significa esto para ti?
El Artículo 5(5) es importante para todas las Instituciones Sanitarias que participan en el desarrollo de dispositivos internos.
Si quieres saber más sobre el Artículo 5(5) y su relación con la nueva ISO 15189:2022 «Laboratorios médicos – Requisitos de calidad y competencia», consulta también nuestro blog.
Ponte en contacto con nuestros expertos
Si quieres hablar de esto más en profundidad con uno de nuestros asesores, no dudes en ponerte en contacto con nosotros.
¿Te ha parecido interesante este artículo? Gracias por compartirlo con tu red:
Aquí encontrarás todas las noticias relacionadas con Regulatory Affairs