
En 2022, se realizaron actualizaciones significativas de las principales normas que afectan a la cualificación de salas blancas. Estas actualizaciones incluyen:
- EU GMP Anexo 1: Fabricación de medicamentos estériles: Este documento describe los requisitos específicos para la fabricación de medicamentos estériles, complementando la Guía EC-GMP.
- ISO 14644-4:2022 Salas blancas y entornos controlados asociados – Parte 4: Diseño, construcción y puesta en marcha: Este documento proporciona un proceso exhaustivo para la creación de salas blancas, que abarca desde los requisitos iniciales hasta el diseño, la construcción y la puesta en marcha.
¿Quieres descubrir las principales actualizaciones? ¿Estás interesado en conocer los requisitos esenciales para la cualificación de salas blancas? En este artículo se describen las principales etapas del ciclo de vida de las salas blancas, con especial atención a la importancia de emplear una metodología exhaustiva de evaluación de riesgos.
¿Qué es una sala blanca?
Según la referencia normativa ISO 14644-1:2015, una sala blanca es un entorno controlado en el que se gestiona y clasifica la concentración de partículas en suspensión en el aire. Su diseño, construcción y funcionamiento controlan eficazmente la introducción, generación y retención de partículas en el interior de la sala. Además, otros parámetros importantes como la temperatura, la humedad y la presión se controlan según sea necesario.
¿Qué es la cualificación de salas blancas?
De acuerdo con las directrices de la EU-GMP, las instalaciones deben estar estratégicamente ubicadas, diseñadas, construidas, adaptadas y mantenidas en consonancia con las operaciones previstas. La disposición y el diseño deben dar prioridad a minimizar el riesgo de errores y facilitar procesos eficaces de limpieza y mantenimiento para evitar la contaminación cruzada, la acumulación de polvo o suciedad y cualquier posible impacto negativo en la calidad del producto.
Además, el anexo 1 del manual GMP de la UE establece específicamente que las salas blancas y los equipos de aire limpio deben someterse a procedimientos de cualificación siguiendo la metodología descrita en el anexo 15. Destaca la importancia de distinguir claramente la cualificación de las salas blancas, incluida la clasificación, de la vigilancia ambiental operativa.
WHITEPAPER GRATUITO
Cualificación de nuevas instalaciones GMP
En este whitepaper, profundizamos en las complejidades
de establecer una nueva instalación GMP. Nos centramos en
posibles obstáculos y las mejores prácticas para proporcionar una
visión conjunta.

Actividades de cualificación de salas blancas
Así pues, las actividades de cualificación de salas blancas deben tener en cuenta todas las etapas, desde el desarrollo inicial de la especificación de los requisitos del usuario hasta el final del uso de la instalación (ver la figura 1).
Se indican las principales etapas y algunos criterios sugeridos (aunque dependen de las circunstancias de cada proyecto y pueden ser diferentes) que podrían incluirse en cada etapa (Anexo 15):
- Especificación de requisitos de usuario (URS),
- Cualificación del diseño (DQ),
- Pruebas de aceptación en fábrica (FAT) / Pruebas de aceptación in situ (SAT),
- Cualificación de la instalación (IQ),
- Cualificación operativa (OQ),
- Cualificación del rendimiento (PQ).
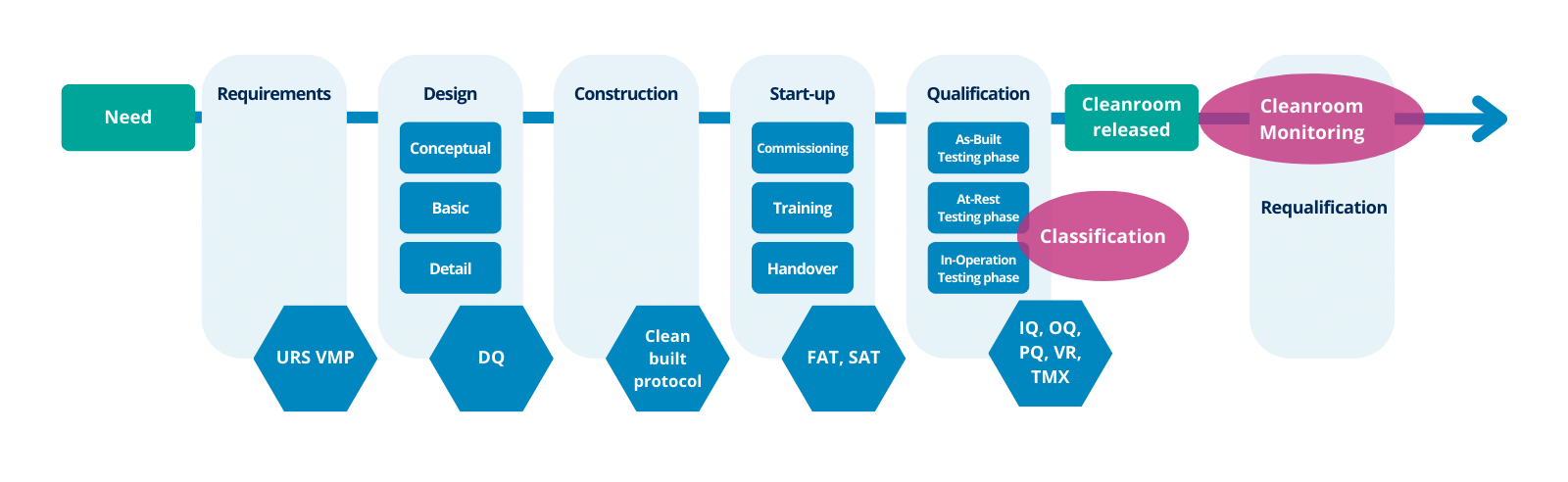
Figura 1 – Cronología de la sala blanca
Especificación de los requisitos de usuario (URS)
El primer paso en el proceso de cualificación es la definición de la Especificación de Requisitos de Usuario (URS). Para ello, se realizará un análisis de la necesidad de una sala blanca y su justificación. Este análisis abordará, entre otras cosas:
- riesgo de contaminación para los productos, los procesos, las personas y el medio ambiente;
- requisitos legales;
- normativa pertinente;
- aspectos relacionados con la empresa (viabilidad financiera y capacidad de recursos);
- y las necesidades futuras.
Cualificación del Diseño (DQ)
El resultado de los requisitos (URS) es la entrada para el diseño. El diseño de la sala limpia deberá tener en cuenta una estrategia eficaz de control de la contaminación en todos los aspectos de su construcción, ensayo, funcionamiento, mantenimiento y ciclo de vida.
Suele haber tres etapas en el proceso general de diseño: diseño conceptual, diseño básico y diseño detallado (ISO 14644-4:2022). Dependiendo de la naturaleza y la escala del proyecto, estas etapas pueden ejecutarse en uno o más pasos con las iteraciones y revisiones de diseño apropiadas.
Al final de cada fase de diseño, se elaborará(n) un(os) documento(s) de diseño acordado(s).
Pruebas de aceptación en fábrica (FAT) / pruebas de aceptación in situ (SAT)
La instalación se construirá de acuerdo con el diseño detallado y las especificaciones acordadas, así como con el plan de construcción y el plan de calidad. Como propone la norma ISO 14644-4:2022, un protocolo de construcción limpia puede apoyar la fase de construcción para limitar las fuentes de contaminación.
Una vez finalizada la construcción de una instalación, el periodo de puesta en marcha comienza con la puesta en servicio para confirmar que la instalación está completa y funciona según lo especificado.
Se llevarán a cabo una serie de verificaciones para demostrar que todos los parámetros de rendimiento se ajustan a la especificación acordada y determinar que todas las partes de la instalación funcionan conjuntamente para alcanzar las condiciones diseñadas.
En el caso de una sala limpia, esto incluirá al menos una prueba de clasificación conforme a la norma ISO 14644-1. Podrán evaluarse otros parámetros, como la contaminación por partículas viables, la contaminación química, etc. Se documentará la justificación del alcance de la verificación. En determinadas condiciones, estas pruebas pueden aprovecharse en las siguientes fases de cualificación (IQ y OQ).
Cualificación de la instalación (IQ)
Este paso podría considerarse una «fase de pruebas as-built«. Una sala limpia «As-built» es una sala limpia funcional y lista para su uso, pero que aún no dispone de equipos ni personal en su interior. Las pruebas as-built son un paso inicial de cualificación de la sala limpia, que refleja la calidad del aire de suministro. De forma no exhaustiva, se pueden realizar las siguientes pruebas: Calibración de HVAC, verificación de bucles P&ID, revisión de datos de pruebas de integridad de filtros HEPA, estado de calibración de equipos críticos,…
En esta fase, también debe medirse la contaminación microbiana del aire y de la superficie para determinar la recuperación inicial (de referencia) de microorganismos.
Cualificación operativa (OQ)
Este paso podría considerarse una fase de «Pruebas en reposo«. Las salas limpias en reposo o «At-rest» están completas, son funcionales y están listas para su uso. Se dispone del equipo, pero no del personal. La realización de pruebas de salas limpias en reposo permite realizar pruebas de humos exhaustivas en superficies de equipos instalados pero que aún no están en funcionamiento.
El protocolo OQ debe abordar, al menos, las siguientes pruebas:
- Instalación de pruebas de estanqueidad e integridad del sistema de filtrado,
- Pruebas de flujo de aire: volumen y velocidad,
- Prueba de diferencia de presión de aire,
- Prueba y visualización de la dirección del flujo de aire,
- Contaminación microbiana aérea y superficial,
- Prueba de medición de la temperatura,
- Prueba de humedad relativa,
- Prueba de recuperación,
- Prueba de fugas de contención.
En esta fase, se puede realizar la clasificación de la sala limpia «en reposo«. En la norma ISO 14644-1 se indica el número mínimo de puntos de muestreo y su ubicación.
Como se solicita en el anexo 1, la clasificación de salas limpias forma parte de la cualificación de salas limpias y es un método de evaluación del nivel de limpieza del aire con respecto a una especificación para una sala limpia o un equipo de aire limpio mediante la medición de la concentración total de partículas.
Para el área de procesamiento aséptico y el entorno de fondo (las áreas de grado A y grado B, respectivamente), se deben considerar ubicaciones adicionales de muestreo y se deben evaluar todas las áreas críticas de procesamiento, como el punto de llenado y las cubetas de alimentación de cierre de envases. Los lugares críticos de procesamiento deben determinarse mediante una evaluación de riesgos documentada y el conocimiento del proceso y las operaciones que se van a realizar en la zona.
La contaminación microbiana del aire y de la superficie también debe medirse «en reposo». El número y la ubicación de los puntos de muestreo deben basarse en una evaluación de riesgos documentada.
Por lo tanto, según el anexo 1, se requieren dos tipos de evaluación de riesgos, una para las partículas y otra para la contaminación microbiana.
Cualificación del rendimiento (PQ)
Este paso podría considerarse una fase de «Pruebas en funcionamiento«. Esta fase se realiza cuando todos los equipos y el personal están en funcionamiento dentro de una sala limpia. Esta prueba final tiene por objeto demostrar que la sala blanca dispone de todas las prestaciones operativas necesarias para una aplicación segura de la sala blanca.
Se puede realizar la clasificación de la sala limpia en estado «En funcionamiento». Para la clasificación en sala limpia, el total de partículas iguales o superiores a 0,5 y 5 μm debe medirse de acuerdo con los límites especificados en la tabla 1 del anexo 1.
El nivel de contaminación microbiana de las salas blancas también debe determinarse «en estado de funcionamiento». El número de ubicaciones de muestreo debe basarse en una evaluación de riesgos documentada y en los resultados obtenidos de las pruebas anteriores (clasificación de salas, estudios de visualización del aire) y en el conocimiento del proceso y las operaciones que se van a realizar en la zona. Los límites máximos de contaminación microbiana durante la calificación para cada grado figuran en el cuadro 2 del anexo 1.
Lanzamiento de la sala blanca
La fase de aceptación y lanzamiento debe realizarse cuando se hayan completado todas las fases de prueba anteriores y se hayan resuelto satisfactoriamente todas las discrepancias. El proceso de aceptación y liberación se completa mediante un informe de síntesis. También puede utilizarse una matriz de trazabilidad como herramienta para identificar el aspecto crítico e incluye referencias a los documentos probados en relación con los requisitos.
Supervisión de salas blancas
Tras su liberación, las salas limpias deben ser objeto de seguimiento. El anexo 1 especifica que la vigilancia ambiental operativa debe diferenciarse claramente de la cualificación de la sala limpia (incluida la clasificación).
El programa de vigilancia operativa del medio ambiente debe contener, según las orientaciones, los siguientes elementos:
- Control medioambiental (partículas totales),
- Control medioambiental y del personal (partícula viable),
- Temperatura, humedad relativa y otras características específicas,
- Simulación de procesos asépticos (sólo para productos fabricados asépticamente).
Explica qué factores deben tenerse en cuenta en la evaluación de riesgos. Entre ellos se incluyen los lugares de muestreo, la frecuencia del seguimiento, los métodos de seguimiento utilizados y las condiciones de incubación.
La información del programa de vigilancia ambiental debe utilizarse para la liberación de productos, pero también para la evaluación continua de las condiciones de las salas blancas y durante las investigaciones.
Las evaluaciones de riesgos deben basarse en el conocimiento de los procesos, así como en los datos de control históricos, los datos de control de cualificación y la propia instalación.
Recualificación
La recualificación de las salas blancas debe realizarse periódicamente siguiendo procedimientos definidos. La recualificación debe incluir como mínimo las siguientes pruebas:
- clasificación de la sala blanca (concentración total de partículas),
- prueba de integridad de los filtros finales,
- medición del caudal de aire,
- verificación de la diferencia de presión de aire entre las habitaciones,
- y prueba de velocidad del aire.
Después de cualquier cambio, también debe llevarse a cabo una recualificación adecuada a través del proceso de gestión de cambios.
Conclusión
Asegúrate de que la cualificación de tu sala blanca cumple las normas EU GMP Anexo 1 e ISO 14644-4:2022 y de que tu evaluación de riesgos está suficientemente justificada.
Las revisiones de ambos reglamentos permitieron aclarar algunas nociones, como la disposición de las distintas fases de las pruebas (cualificación, clasificación, recalificación, control), e hicieron especial hincapié en la noción de riesgo. Así, las normas exigen que cada prueba y plan de pruebas esté justificado por una evaluación de riesgos.
¿Tienes problemas con las actualizaciones? ¿Eres nuevo en la cualificación de salas blancas? ¿O necesitas consejo?
Nuestros expertos estarán encantados de ayudarte con el mejor programa de validación de acuerdo con tus recursos disponibles.No dudes en ponerte en contacto con nosotros si tienes alguna pregunta.
Expert knowledge in Qualification & Validation
- EudraLex – Volumen 4 – Guía sobre las buenas prácticas de fabricación (GMP): Parte 1.
- EU GMP Anexo 1: Fabricación de medicamentos estériles.
- EU GMP Anexo 15: Cualificación y validación.
- ISO 14644-1:2015 Salas blancas y entornos controlados asociados – Parte 1. Clasificación de la limpieza del aire por concentración de partículas: Clasificación de la limpieza del aire por concentración de partículas.
- ISO 14644-4:2022 Salas blancas y ambientes controlados asociados – Parte 4: Diseño, construcción y puesta en marcha.
- ISPE Baseline ® Guías de ingeniería farmacéutica, Vol. 5, Puesta en servicio y cualificación