







.png?width=109&height=108&name=Pharma%20(2).png)
.png?width=111&height=108&name=Medical%20Devices%20(2).png)
.png?width=84&height=107&name=IVD%20(2).png)

What is a cleanroom?
According to the normative reference ISO 14644-1:2015, a cleanroom is a controlled environment where the concentration of airborne particles is managed and classified. It is designed, constructed, and operated in a way that effectively controls the introduction, generation, and retention of particles within the room. Additionally, other important parameters such as temperature, humidity, and pressure are controlled as required.
What is cleanroom qualification?
As per the guidelines outlined in EU-GMP, premises should be strategically located, designed, constructed, adapted, and maintained to align with the intended operations. The layout and design should prioritize minimizing the risk of errors and facilitating effective cleaning and maintenance processes to prevent cross-contamination, accumulation of dust or dirt, and any potential negative impact on product quality.
Furthermore, the EU GMP Annex 1 specifically mandates that cleanrooms and clean air equipment must undergo qualification procedures following the methodology outlined in Annex 15. It emphasizes the importance of clearly distinguishing cleanroom qualification, including classification, from operational environmental monitoring.

Cleanroom qualification activities
So, qualification activities of cleanrooms should consider all stages from the initial development of the user requirements specification through to the end of the use of the facility (See figure 1).
The main stages and some suggested criteria (although this depends on individual project circumstances and may be different) which could be included in each stage are indicated (Annex 15):
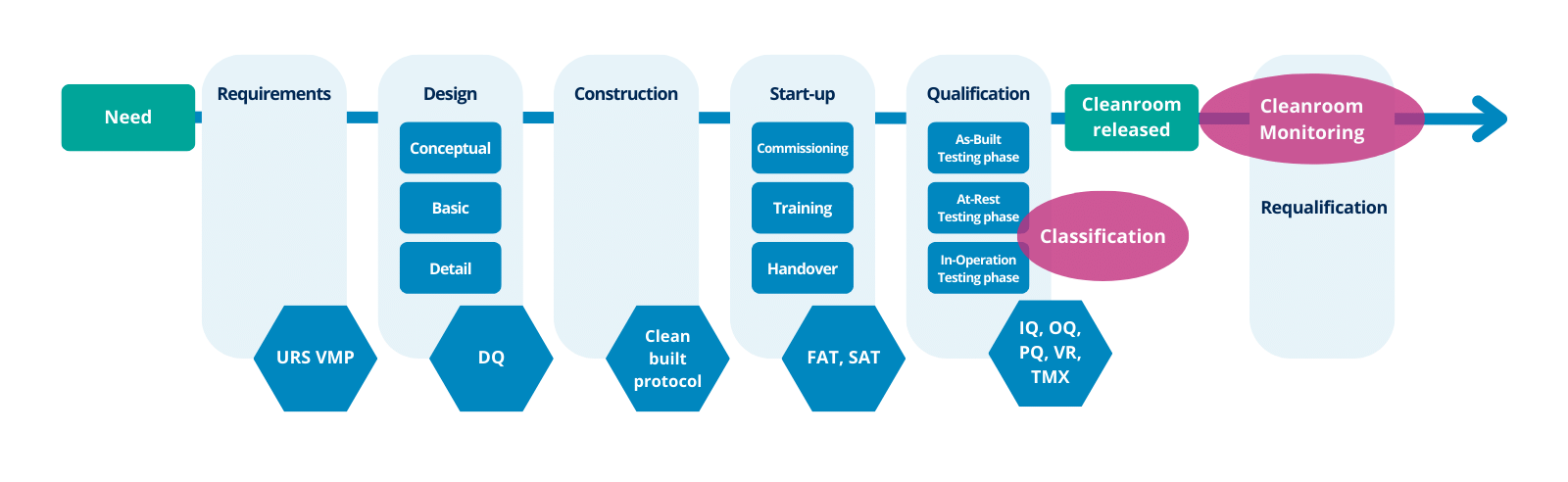
Cleanroom monitoring
After their release, the cleanrooms should be monitored. Annex 1 specifies that operational environmental monitoring should be clearly differentiated from cleanroom qualification (including classification).
The program of operational environmental monitoring should, according to the guidance, contain the following elements:








