

Procedimiento nacional
Extensiones de línea
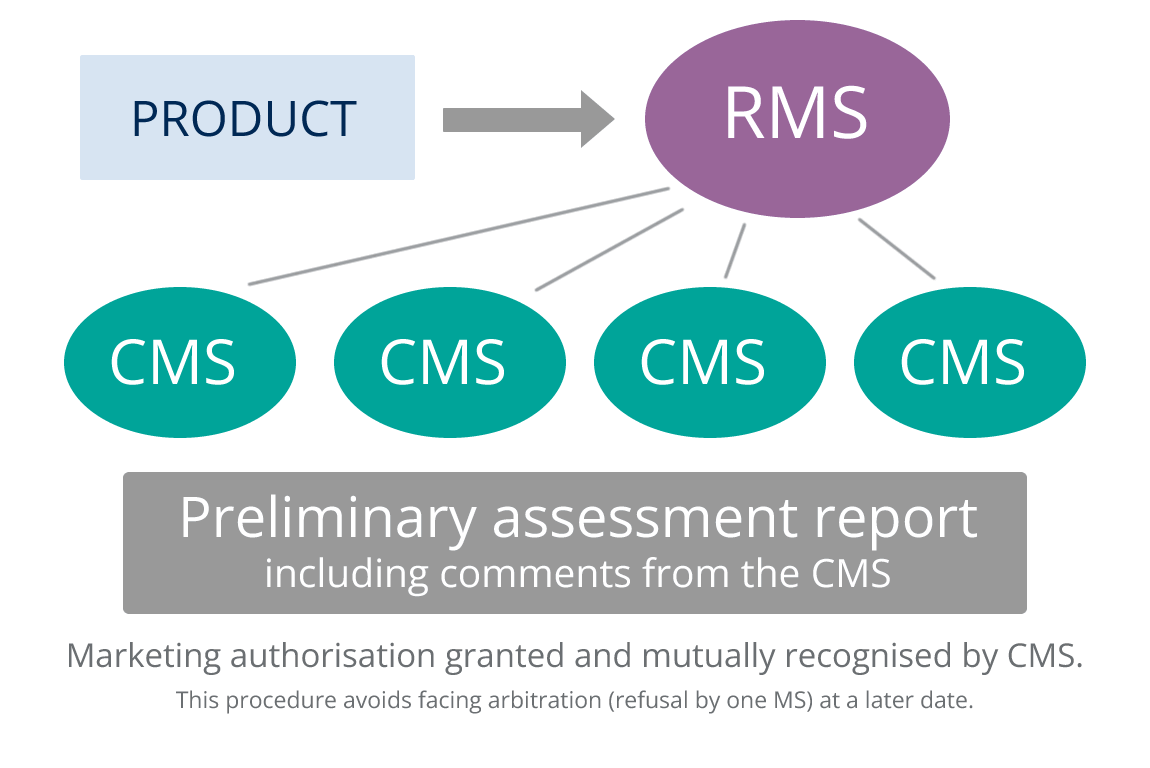
Procedimiento de reconocimiento mutuo (PRM)
Procedimiento descentralizado
El Procedimiento Descentralizado (DCP), en general, sigue los mismos principios del MRP. Una licencia aprobada en un Estado miembro (EM) debe ser reconocida mutuamente en otros Estados miembros (EM), suponiendo que los criterios de evaluación en los Estados miembros de la UE estén suficientemente armonizados y tengan el mismo nivel. El DCP debe utilizarse si el objetivo es obtener una autorización de comercialización en varios Estados miembros (EM) cuando, en el momento de la solicitud, el medicamento en cuestión aún no haya recibido una autorización de comercialización en ningún EM. La principal diferencia entre el MRP y el DCP radica en que los Estados Miembros Interesados (EMI) en un DCP participan al inicio del procedimiento, a diferencia de esperar a la aprobación antes de presentar una solicitud en el EMI.
Procedimiento centralizado
