
In June 2022, concerns were expressed that severe challenges related to the implementation of Regulation (EU) 2017/745 on medical devices (MDR) would threaten the continued availability of several medical devices on the EU market. The biggest challenges reported were the insufficient capacity of notified bodies to certify medical devices in accordance with MDR and the level of preparedness of the manufacturers by the end of the transition period, set on the 26th of May 2024.
In several meetings around this topic later on in 2022, more concerns were raised by multiple stakeholders, and the request was made to extend the timelines of the transition period. As a response to these concerns, the Council of the European Union sent out a communication on the 6th of December 2022 around the implementation of the MDR.
What has changed about the MDR transition and implementation since Dec. 6, 2022?
The MDR transition period with staggered deadlines depending on the risk class of the device was extended to December 31, 2027, for class IIb implantable and class III devices, and to December 31, 2028, for class I sterile/measuring and MDD class I self-certified devices, class IIa and all other class IIb devices, for those devices that need involvement in the conformity assessment by the notified bodies.
For legal and practical reasons, the extension of this transitional period could be combined with an extension of the validity of the certificates issued under the Medical Devices Directives earlier.
This extension applies only to devices that do not present an unacceptable risk to the health and safety of the patients and/or users, that have not undergone significant changes in design or intended purpose AND for which the manufacturers have already undertaken the necessary steps to launch the certification process under the MDR (see the next paragraph for details).
On March 15th, 2023, Regulation (EU) 2023/607 was published, outlining the extension to the MDR transitional provisions. Guidance on how to implement is expected to follow soon.
What does this mean for your medical device?
If your medical device is currently in a pre-market phase, it will have to be classified according to MDR and its technical file should comply with all requirements as laid out in this legislation. The above-mentioned MDR transition deadlines do not apply in this case. Once all requirements are met, you can submit your technical file for CE marking as usual.
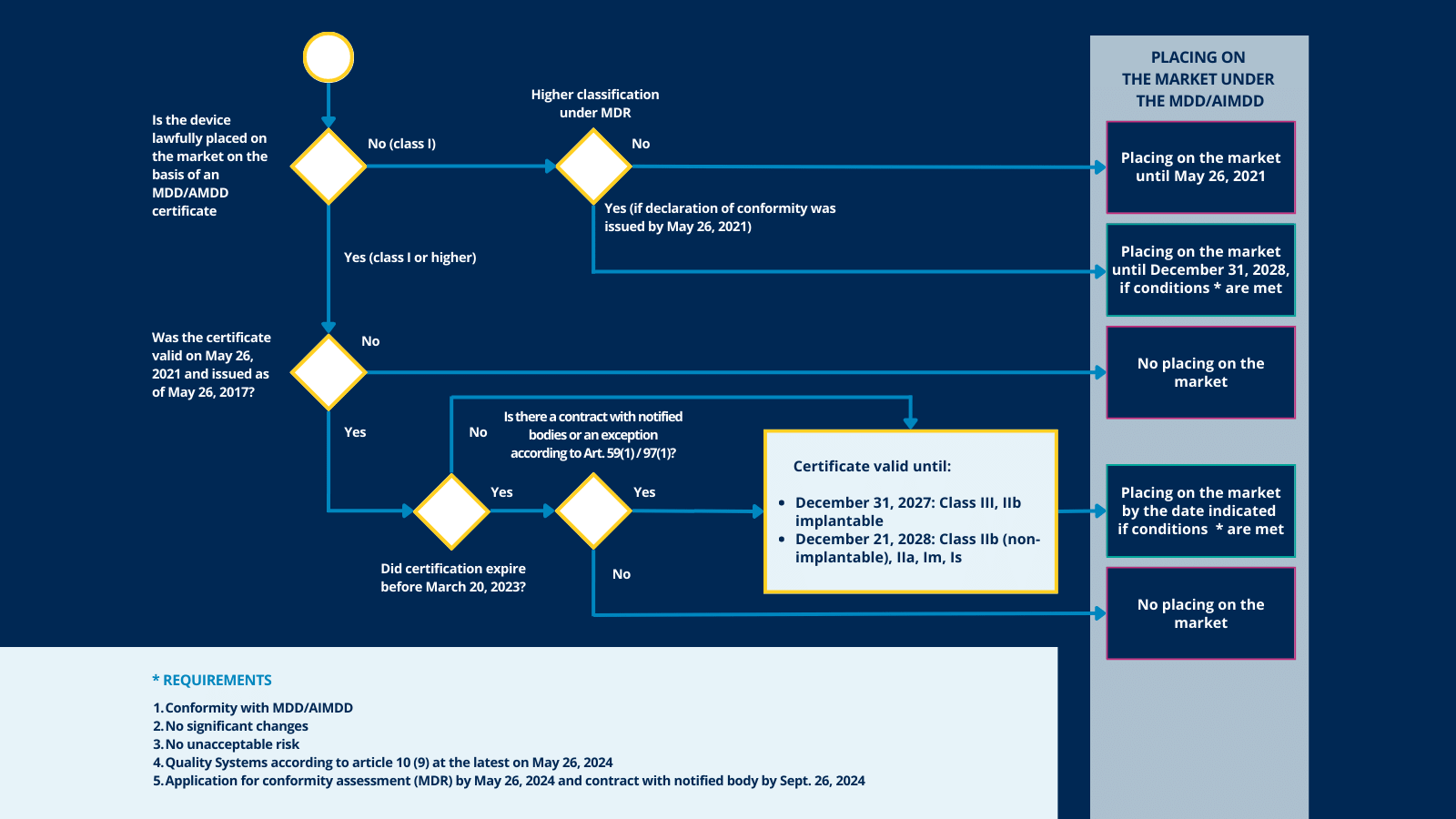
If your device has been on the market and was previously certified under the MDD before May 26, 2021, the following applies:
1. Check the expiration date of the current certificate.
If the certificate was still valid on 26MAY21 (and not withdrawn) but expired before 20MAR23, a written agreement must be in place from before this date, that the certificate was extended (signed by the manufacturer and the notified body, or a derogation from the applicable competent authority).
If still valid after March 20, 2023, the certificate is extended automatically, depending on the classification of the device, to December 31, 2027, or 2028, BUT under the following conditions:
- The device still complies with the MDD;
- There are no significant changes in the design and intended purpose;
- The devices do not present an unacceptable risk to the health and safety of the patients or users;
- No later than May 26, 2024, the manufacturer has put in place a quality management system in accordance with MDR;
- No later than May 26, 2024, the manufacturer has lodged a formal application with a notified body for a conformity assessment.
2. Check the classification of your device
Check the classification of your device and determine if this is still valid under MDR or whether your device falls under a higher-risk class.
In case the device falls under a higher class, there should be no delay in starting to prepare the technical file for MDR transitioning, as more strict requirements might apply.
3. Decide on the continuation of the device on the market
Decide on the continuation of the device on the market after May 26, 2024, and in what timeframe these would be taken off the market if discontinued. The decision and timeframe of discontinuation should be communicated to the healthcare providers in due time.
4. Commence the update of the technical file
Commence the update of the technical file to meet the high requirements and expectations per the MDR, as this is a condition written in the latest communication of the Council of the European Union.
5. Perform a systemic literature review
Perform a systemic literature review, certainly to describe the state of the art of the medical field of your device, and to define the benchmark safety and performance parameters to be able to assess the benefit/risk ratio for your device, and in comparison to the state of the art. This literature review will also identify the gaps in clinical data, required for your device to be re-certified under MDR.
6. Write or update the Clinical Evaluation Plan
Write or update the Clinical Evaluation Plan (CEP), as this is an important aspect of the technical file, including the clinical development plan.
For existing and legacy devices, certification might have been done without clinical data gained through clinical investigations. For class IIb implantable and class III devices, these data are now required per MDR and might imply the need for post-market clinical follow-up studies.
As these studies take time, you want to start immediately to be able to comply with the deadline of transitioning, even with the extended period.
FREE WHITEPAPER
Clinical evaluation for medical devices under MDR
In this whitepaper, we’ll guide you through crucial regulatory documents (CEP, CER, etc.) pertaining to the clinical evaluation process of your medical device.

How can QbD Clinical help you?
At QBD Clinical, we have a dedicated team of brilliant medical writers with experience in clinical evaluations and other technical documents like e.g., PMCFU plans and reports, and requirements of MDR towards clinical investigations. From experience in the past years, we know that literature review (updates) and clinical evaluations take up to 6 months or more, hence, the extension period merely gives you more time to do this properly, rather than delaying the process.
QbD Clinical also hosts a team of project managers and CRAs to manage your clinical investigations or post-market clinical follow-up studies, where required (i.e., when a gap in clinical data is defined by the literature review and clinical evaluation plan).
QbD Regulatory can assist in the completion of other aspects of the technical files, like. e.g., risk management plans and reports, post-market surveillance, Instructions for Use, etc.
Experts in the field of MDR recommend continuing with the completion of your technical files and providing the notified bodies with timely dossiers for review, to avoid issues with continuing the certification of your device in a timely manner. Although notified bodies have been ramping up capacity, there is no endlessness in processing the applications and one would need to avoid queueing before the extended deadlines again. This is the only way to assure that certifications will be given in time to avoid shortages of medical devices in the market and the associated risk of a public health crisis.
A Q&A on the extension of the MDR transition period has just been published by the European Commission.
For help with any of the mentioned documents and beyond, QbD Clinical and Regulatory can provide a solution. Don’t hesitate to contact us.