.png?width=109&height=108&name=Pharma%20(2).png)







.png)
Marco normativo actual de la UE para titulares de MIA

Quercus, su laboratorio GMP de confianza
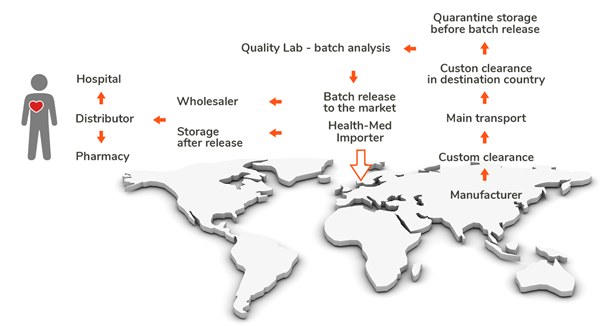
Quercus Laboratories es miembro del grupo QbD y es un laboratorio GMP de confianza que apoya a la industria farmacéutica con una amplia gama de servicios de pruebas de control de calidad. Ofrecemos un servicio completo de pruebas de control de calidad químicas y microbiológicas de materias primas y productos farmacéuticos acabados.
Nuestras capacidades incluyen:
Validación de métodos analíticos - Liberación de lotes - Servicios de importación a la UE - Transferencias técnicas de métodos analíticos - Pruebas de lotes y certificados de análisis - Certificación de plantas de fabricación para el cumplimiento de las GMP de la UE - Revisión documental - Certificado de lote para liberación - Servicios de respaldo de QP - Estudios de estabilidad adicionales - Almacenamiento seguro de muestras de retención - Titular de MA - Apoyo en asuntos regulatorios: Redacción de expedientes CTD y solicitud de autorización de comercialización (CP/DCP/MRP/NP).