

Actividades de cualificación de salas limpias
Así pues, las actividades de cualificación de las salas blancas deben considerar todas las etapas, desde el desarrollo inicial de la especificación de los requisitos del usuario hasta el final del uso de la instalación (véase la figura 1).
Se indican las principales etapas y algunos criterios sugeridos (aunque esto depende de las circunstancias de cada proyecto y puede ser diferente) que podrían incluirse en cada etapa ( Anexo 15):
- Especificación de los requisitos del usuario (URS),
- Cualificación del diseño (DQ) ,
- Pruebas de aceptación en fábrica (FAT) / Pruebas de aceptación in situ (SAT) ,
- Cualificación de la instalación (IQ) ,
- Cualificación del funcionamiento (OQ) ,
- Cualificación de la ejecución del proceso (PQ).
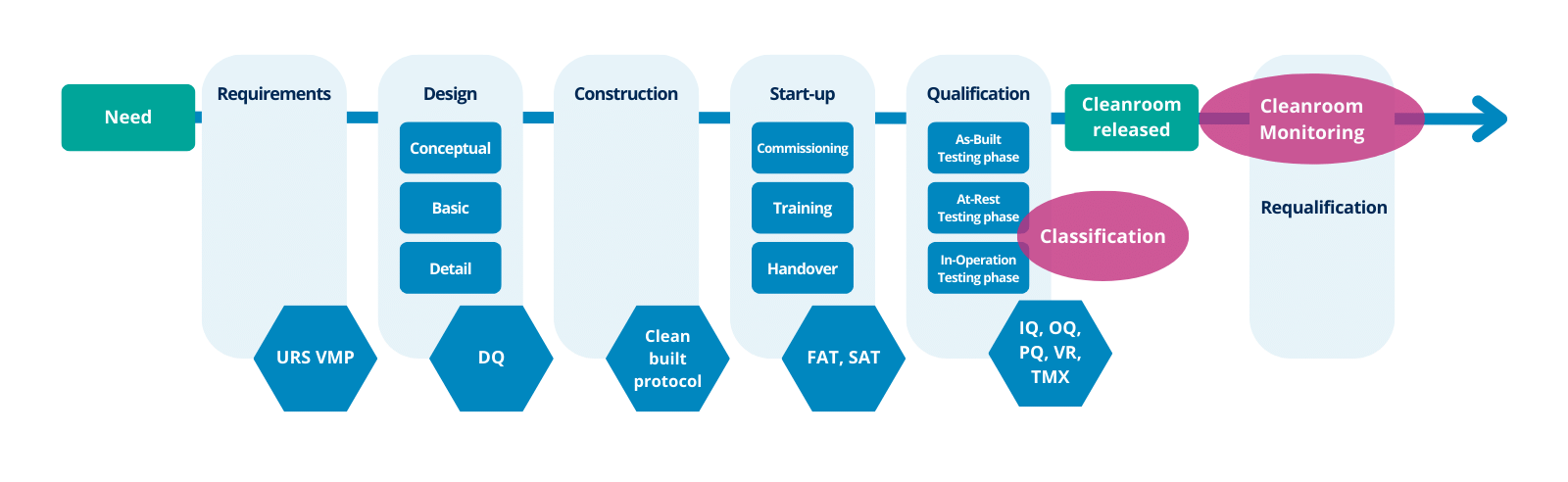
Figura 1 - Cronología de la sala blanca
Especificación de los requisitos de usuario
El primer paso en el proceso de cualificación es la definición de la especificación de requisitos de usuario (URS). Para ello, se realizará un análisis de la necesidad de una sala limpia y su justificación. Este análisis abordará, entre otros:
- riesgo de contaminación para productos, procesos, personas y medio ambiente;
- requisitos legales;
- la normativa pertinente;
- aspectos relacionados con el negocio (viabilidad financiera y capacidad de recursos);
- y necesidades futuras.
Cualificación del Diseño (DQ)
La salida de los requisitos (URS) es la entrada para el diseño. El diseño de la sala limpia debe considerar una estrategia efectiva de control de la contaminación para todos los aspectos de su construcción, pruebas, operación, mantenimiento y ciclo de vida.
Por lo general, hay tres etapas en el proceso de diseño general: diseño conceptual, diseño básico y diseño detallado ( ISO 14644-4:2022). Dependiendo de la naturaleza y la escala del proyecto, estas etapas se pueden ejecutar en uno o más pasos con iteraciones y revisiones de diseño adecuadas.
Al final de cada etapa de diseño, se elaborará un documento o documentos de diseño acordados.
Pruebas de aceptación en fábrica (FAT) / Pruebas de aceptación en el sitio (SAT)
La instalación se construirá de acuerdo con el diseño detallado y las especificaciones acordadas, así como con el plan de construcción y el plan de calidad. Tal y como propone la norma ISO 14644-4:2022, un protocolo de construcción limpia puede apoyar la fase de construcción para limitar las fuentes de contaminación.
Una vez finalizada la construcción de una instalación, el período de puesta en marcha comienza con la puesta en marcha para confirmar que la instalación está completa y funciona según lo especificado.
Se llevarán a cabo una serie de verificaciones para demostrar que todos los parámetros de rendimiento se ajustan a las especificaciones acordadas y para determinar que todas las partes de la instalación funcionan juntas para alcanzar las condiciones diseñadas.
En el caso de una sala limpia, deberá incluir, como mínimo, un ensayo de clasificación de conformidad con la norma ISO 14644-1. Se pueden evaluar otros parámetros, como la contaminación por partículas viables, la contaminación química y... Se documentarán los motivos del alcance de la verificación. Bajo ciertas condiciones, estas pruebas se pueden aprovechar en las siguientes fases de cualificación (IQ y OQ).
Cualificación de la Instalación (IQ)
Este paso podría considerarse como una "fase de prueba As-Built". Una sala limpia As-built se refiere a una sala limpia que es funcional y está lista para funcionar, pero que aún no tiene ningún equipo ni personal en su interior. Las pruebas as-built son un paso inicial de calificación de la sala limpia, que refleja la calidad del aire de suministro. De forma no exhaustiva, se pueden realizar las siguientes pruebas: calibración de HVAC, verificación de bucle P&ID, revisión de datos de pruebas de integridad de filtros HEPA, estado de calibración de equipos críticos, ...
En esta etapa, también se debe medir la contaminación microbiana en el aire y en la superficie para determinar la recuperación inicial (de referencia) de los microorganismos.
Cualificación del Funcionamiento (OQ)
Este paso podría considerarse una fase de "Pruebas en reposo". Las salas blancas en reposo están completas, funcionales y listas para usar. El equipo está en su lugar, pero no el personal. La realización de pruebas de sala limpia en reposo permite realizar pruebas exhaustivas de humo de las superficies de los equipos que están instalados pero que aún no funcionan.
El protocolo OQ debe abordar, al menos, las siguientes pruebas:
- Pruebas de integridad y fugas del sistema de filtro instalado,
- Pruebas de flujo de aire: volumen y velocidad,
- Prueba de diferencia de presión de aire,
- Prueba y visualización de la dirección del flujo de aire,
- Contaminación microbiana en el aire y la superficie,
- Prueba de medición de temperatura,
- Prueba de humedad relativa,
- Prueba de recuperación,
- Prueba de fugas de contención.
En esta etapa, se puede realizar la clasificación de la sala limpia "en reposo". El número mínimo de ubicaciones de muestreo y su posicionamiento se pueden encontrar en la norma ISO 14644-1.
Tal como se solicita en el Anexo 1, la clasificación de salas limpias forma parte de la calificación de salas limpias y es un método para evaluar el nivel de limpieza del aire en relación con una especificación para una sala limpia o un equipo de aire limpio mediante la medición de la concentración total de partículas.
Para el área de procesamiento aséptico y el entorno de fondo (las áreas de grado A y grado B, respectivamente), se deben considerar ubicaciones de muestras adicionales y se deben evaluar todas las áreas críticas de procesamiento, como el punto de llenado y los recipientes de alimentación de cierre del contenedor. Las ubicaciones críticas de procesamiento deben determinarse mediante una evaluación de riesgos documentada y el conocimiento del proceso y las operaciones que se realizarán en el área.
La contaminación microbiana en el aire y en la superficie también debe medirse tanto "en reposo". El número y la ubicación de los puntos de muestreo deben basarse en una evaluación de riesgos documentada.
Por lo tanto, de acuerdo con el Anexo 1, se requieren dos tipos de evaluación de riesgos, uno para las partículas y otro para la contaminación microbiana.
Cualificación de la ejecución del proceso (PQ)
Este paso podría considerarse una fase de "Pruebas en Operación". Esta fase se lleva a cabo cuando todo el equipo y el personal están en funcionamiento dentro de una sala limpia. Esta prueba final tiene como objetivo demostrar que la sala limpia tiene todo el rendimiento operativo requerido para una aplicación segura de sala limpia.
Se puede realizar la clasificación de la sala limpia en el estado "En funcionamiento". Para la clasificación de salas limpias, el total de partículas iguales o superiores a 0,5 y 5 μm debe medirse de acuerdo con los límites especificados en el cuadro 1 del Anexo 1.
El nivel de contaminación microbiana de las salas limpias también debe determinarse en estados "en funcionamiento". El número de lugares de muestreo debe basarse en una evaluación de riesgos documentada y en los resultados obtenidos de las pruebas anteriores (clasificación de salas, estudios de visualización del aire) y en el conocimiento del proceso y las operaciones que se van a realizar en la zona. Los límites máximos de contaminación microbiana durante la calificación para cada grado se indican en la Tabla 2 del Anexo 1.
Liberación de la sala blanca
La fase de aceptación y liberación debe realizarse cuando se hayan completado todas las fases de prueba anteriores y se hayan cerrado con éxito todas las discrepancias. El proceso de aceptación y liberación se completa a través de un informe resumido. Una matriz de trazabilidad también se puede utilizar como herramienta para identificar el aspecto crítico e incluye referencias a los documentos probados en relación con los requisitos.
Supervisión de salas blancas
Tras su puesta en funcionamiento, las salas limpias deben ser objeto de seguimiento. El anexo 1 especifica que la vigilancia ambiental operativa debe diferenciarse claramente de la cualificación de la sala limpia (incluida la clasificación).
El programa de vigilancia ambiental operativa debe contener, de acuerdo con las orientaciones, los siguientes elementos:
- Monitorización ambiental (partículas totales),
- Control ambiental y personal (partículas viables),
- Temperatura, humedad relativa y otras características específicas,
- Simulación del proceso aséptico (sólo para productos fabricados asépticamente).
Explica qué factores deben tenerse en cuenta en la evaluación de riesgos. Entre ellos se incluyen los lugares de muestreo, la frecuencia de la vigilancia, los métodos de vigilancia utilizados y las condiciones de incubación.
La información del programa de vigilancia ambiental debe utilizarse para la liberación de productos, pero también para la evaluación continua de las condiciones de las salas blancas y durante las investigaciones.
Las evaluaciones de riesgos deben basarse en el conocimiento de los procesos, así como en los datos históricos de vigilancia, los datos de vigilancia de cualificación y la propia instalación.
Recualificación
La recualificación de las salas blancas debe realizarse periódicamente siguiendo procedimientos definidos. La recualificación debe incluir como mínimo las siguientes pruebas:
-
clasificación de la sala limpia (concentración total de partículas)
-
prueba de integridad de los filtros finales,
-
medición del volumen de flujo de aire,
-
verificación de la diferencia de presión de aire entre salas,
-
y prueba de velocidad del aire.
También debe llevarse a cabo una recualificación adecuada después de cualquier cambio a través del proceso de gestión de cambios.
Conclusión
Asegúrate de que la cualificación de su sala blanca cumple la nueva versión de las normas EU GMP Anexo 1 e ISO 14644-4:2022 y de que su evaluación de riesgos está suficientemente justificada.
Las revisiones de ambas normas han permitido aclarar algunas nociones, como la disposición de las distintas fases de ensayo (cualificación, clasificación, recalificación, seguimiento), y han hecho especial hincapié en la noción de riesgo. Así, la nueva versión de las normas exige que cada prueba y cada plan de prueba estén justificados por una evaluación de riesgos.
¿Tienes problemas con las actualizaciones? ¿Eres nuevo en la cualificación de salas blancas? ¿O necesitas asesoramiento?
Nuestros expertos estarán encantados de ayudarte con el mejor programa de validación de acuerdo con tus recursos disponibles

La Validación y la Cualificación son esenciales para garantizar que tus sistemas rindan sin ningún fallo.
Descubre nuestras soluciones.
Sobre el autor
QbD Group
¿Listo para acelerar tu proyecto en Life Sciences? Habla con nuestros expertos.
Contacta con un experto →

