.png?width=109&height=108&name=Pharma%20(2).png)
.png?width=111&height=108&name=Medical%20Devices%20(2).png)
.png?width=84&height=107&name=IVD%20(2).png)





.png)
In recent years, medicinal products for the EU market have been increasingly manufactured outside the EU. While this trend is particularly noticeable in the production of active ingredients, it is also becoming more prevalent in the manufacture of finished medicinal products.
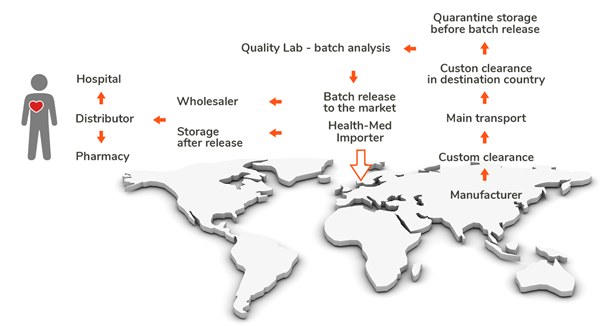
At the same time, globalization has made pharmaceutical supply chains more complex. To ensure oversight, the EU classifies medicine importers as manufacturers, requiring them to obtain a Manufacturing Import Authorization (MIA) and comply with Good Manufacturing Practice (GMP) regulations.
This framework mandates the implementation of a pharmaceutical quality system, adequate staffing and facilities, complaint and recall management, and strict supply chain control.
In this guide, we will explore the key aspects of GMP Annex 21, its implications for medicine importers, and best practices to ensure compliance.
GMP Annex 21: overview
GMP Annex 21 establishes GMP requirements for MIA holders importing medicinal products from outside the EU/EEA. The annex applies to:
- medicinal products for human use
- medicinal products for veterinary use
- investigational medicinal products
However, it does not cover products without an EU/EEA marketing authorization that are directly re-exported.
Defining 'import' under GMP Annex 21
The Annex provides a common interpretation of import, defining it as the physical introduction of a medicinal product from outside the EU/EEA.
Importation is a regulated process, and batch release by a Qualified Person (QP) can only take place after physical importation and customs clearance within an EU/EEA state.
Key sites in the importation process
GMP Annex 21 highlights two critical sites involved in the importation of medicinal products:
- Site of physical import – The location where the medicinal product first enters the EU.
- Site of QP certification – Where a Qualified Person (QP) performs batch certification before release.
These sites must be considered in the broader context of the entire supply chain, which also involves:
-
API manufacturers
-
Third-country medicinal product manufacturers
-
Transport companies
-
QC testing facilities for imported products
-
Marketing Authorization Holders (MAH)