Los países dependen del uso de dispositivos médicos para lograr la cobertura sanitaria universal, responder a las emergencias sanitarias y mantener la seguridad de la población. Vemos cómo las empresas diseñan y desarrollan cada vez más dispositivos y tratan de encontrar su camino hacia la aprobación del mercado. Entre otras cosas, esta estrategia de mercado tiene que tener en cuenta aspectos importantes como lanorma ISO13485 2016, el marcado CE del producto, la aprobación de la FDA y muchos más. Y como el nuevo Reglamento de Productos Sanitarios (MDR ) entrará en vigor en 2021, el proceso se vuelve aún más complejo. No es de extrañar que todas las partes interesadas en los dispositivos médicos quieran subirse a este tren que se aproxima.
El MDR (UE) 2017/745 se acerca rápidamente
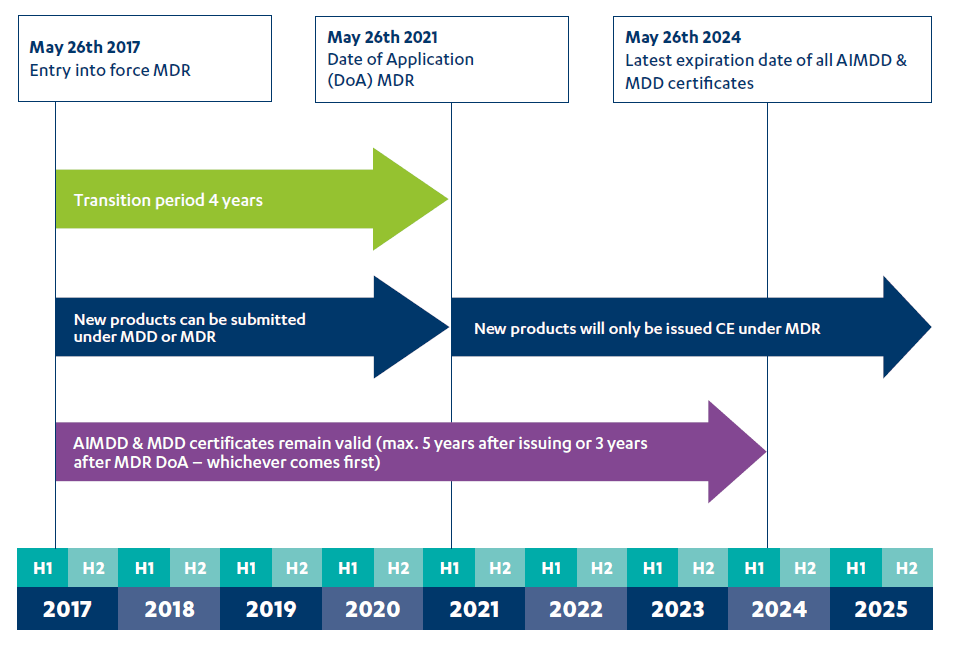
Aunque parezca estar lejos, el tren se acerca y viene rápido. La nueva MDR (UE) 2017/745 sustituirá a la actual Directiva de Productos Sanitarios (MDD) 93/42/CEE y a la Directiva de Productos Sanitarios Implantables Activos (AIMDD) 90/385/CEE. Estas directivas llevan más de 25 años en vigor y han sido la base del mercado único de productos sanitarios en Europa. También definieron el camino hacia la certificación CE. Sin embargo, el excesivo margen de interpretación, las innovaciones tecnológicas y algunos escándalos ilustres han demostrado la necesidad de una normativa más estricta y actualizada. El MDR entró en vigor el 26 de mayo de 2017, iniciando un periodo de transición de tres años. La fecha de aplicación (DoA ) del MDR es el 26 de mayo de 2021. Para entonces, todos los fabricantes de productos sanitarios deberán cumplirla.
Los actores importantes y los fabricantes de productos sanitarios con experiencia están haciendo esfuerzos significativos para estar totalmente preparados para la transición y para recertificar todos sus productos en el mercado antes de la DoA. Sin embargo, las empresas de nueva creación que quieren entrar en el mercado con sus nuevos productos en los próximos años, se enfrentan a la decisión de si seguir presentando la solicitud bajo la MDD o presentarla bajo la más estricta MDR. El momento de la entrada en el mercado es crucial: las presentaciones posteriores al 26 de mayo de 2021 ya no serán aceptadas por los organismos notificados en virtud de la MDD. La disponibilidad de recursos de calidad y reglamentarios, así como el plan estratégico de negocio, determinarán en gran medida la decisión de obtener o no la certificación antes de este plazo.
Cumplir el plazo de transición: dos opciones
Basándose en las consideraciones anteriores, los “nuevos” agentes del mercado de productos sanitarios tienen dos opciones a la hora de presentar sus solicitudes antes del plazo de transición.
- Presentación de solicitudes con arreglo a la Directiva 93/42/CEE: ¿sólo para temerarios?
Esta estrategia es una opción si su dispositivo entra en una clasificación de mayor riesgo según el MDR. La clasificación sigue siendo mayoritariamente la misma en el MDR, pero algunas definiciones y principios básicos implican cambios importantes. Por ejemplo, los instrumentos quirúrgicos reutilizables pertenecerán a una clase de riesgo mayor y requerirán una auditoría de un organismo notificado antes de la aprobación del mercado. Además, las normas para el software como producto sanitario han cambiado. Mientras que la mayoría de las aplicaciones de software se clasifican en la clase I de bajo riesgo, el software bajo el MDR puede entrar en cualquier clase de riesgo, siendo ahora la clase I la excepción. Por lo tanto, es muy recomendable hacer una evaluación exhaustiva de la clasificación actual y futura de su(s) dispositivo(s). Una clasificación de mayor riesgo implica más datos (clínicos), más mecanismos de control y un proceso de evaluación de la conformidad más complejo. Esto hace que la idea de una presentación bajo la MDD sea una opción tentadora. Sin embargo, tenga en cuenta que el tiempo se acaba. Los organismos notificados ya están cerrando las puertas a las auditorías en el marco de la MDD. Cuanto mayor sea la clasificación de riesgo, antes deberá presentar su expediente técnico. Los dispositivos de menor riesgo deben presentarse antes del verano de 2019 para obtener la certificación. En resumen, la presentación de la MDD puede hacerse, pero las empresas deben tener en cuenta el coste y también el margen de error, en caso de que las malas auditorías u otros obstáculos inesperados les impidan cumplir el plazo.
Presentación bajo el MDR 2017/745: la opción más segura
Los primeros organismos notificados serán designados para el MDR a partir del primer trimestre de 2019. Esto significa que ya es posible presentar solicitudes en el marco del MDR europeo. Sin embargo, muchos de los principales organismos notificados no serán designados para el MDR hasta algún momento entre junio y octubre de 2019. Una vez que esto ocurra, los organismos notificados de la UE seguramente dirigirán más sus recursos hacia el tsunami de solicitudes MDR que se producirá a partir de entonces.
Como empresa, no le preocupará el duro plazo de transición, sino sobre todo la fecha de entrada en el mercado que prefiera. Así se gana algo de tiempo, pero hay que tener en cuenta que el archivo de presentación no es el destino final del proceso. Tendrá más tiempo para reunir todas las pruebas clínicas para su Informe de Evaluación Clínica y para desarrollar planes adecuados de Vigilancia Post-Mercado (PMS ) y de Seguimiento Clínico Post-Mercado (PMCF). Además, su Sistema de Gestión de la Calidad ( SGC ) puede adaptarse por completo a los requisitos de la norma ISO13485:2016 (cuya fecha límite es marzo de 2019). Se trata de una importante inversión de recursos y tiempo, pero por una buena causa. Las empresas que cumplan con la MDD deberán finalmente cumplir con el MDR a más tardar en mayo de 2024, ya que todos los certificados MDD emitidos antes de mayo de 2021 expirarán a partir de ese mes. Las empresas de nueva creación que hayan hecho la transición antes, adquirirán más experiencia con el PMCF y el PMS y el Identificador Único de Dispositivos (UDI). Otra ventaja es que las partes interesadas y los clientes confiarán más en su empresa y en sus productos, incluida su seguridad y trazabilidad, cuando ya estén disponibles en la base de datos obligatoria de EUDAMED.
Mi conclusión: Si su empresa tiene los recursos disponibles, si ha reunido suficientes datos clínicos y el modelo de negocio permite la entrada en el mercado antes de mayo de 2021, es prudente empezar a presentar sus dispositivos bajo el MDR 2017/745. Si el contexto de su empresa hace que la presentación de la MDD sea la opción preferida, el esfuerzo simplemente se pospone. Por último, pero no menos importante, sea cual sea la opción que elijas, asegúrate de reunir a especialistas y profesionales de calidad a tu alrededor para tomar las decisiones correctas. Al fin y al cabo, estamos hablando de una transición clave en el mundo de los dispositivos médicos. Sería una pena perder el tren.
