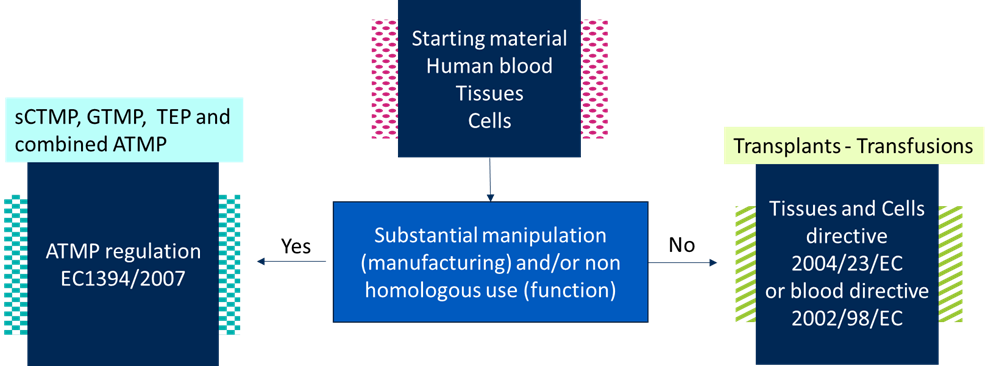
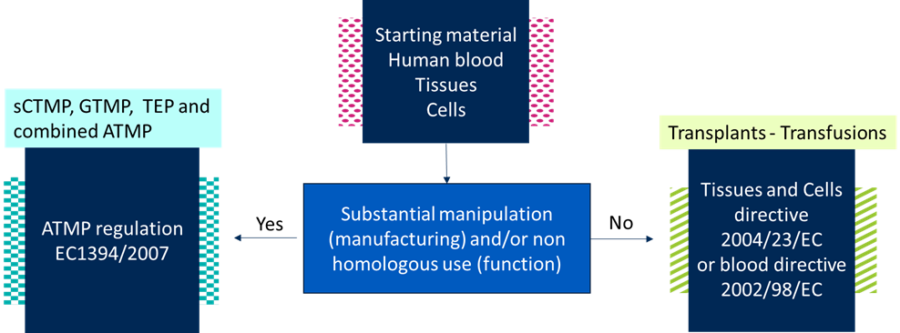
¿Quiere distinguir los materiales de partida utilizados para los ATMP de los utilizados para los trasplantes o las transfusiones? Esta distinción se hace en función de cómo se procesan los materiales de partida y/o se utilizan clínicamente. Hemos enumerado los pasos para hacer tal distinción, con la ayuda de un diagrama de flujo.
Los flujos de trabajo actuales del ATMP comienzan con la selección de donantes y la obtención de material de partida autólogo (es decir, del paciente), alogénico (es decir, de un donante humano) o xenogénico (es decir, de un animal). Estos materiales de partida incluyen la sangre (humana), los tejidos y las células.
La obtención, la manipulación y el paciente/donante Las diferencias específicas de los materiales de partida tienen un impacto significativo en la proceso de fabricación. Por lo tanto, la variabilidad de las materias primas es un factor de entrada que hay que tener en cuenta durante el desarrollo del producto y de fabricación. Un conocimiento profundo de los atributos críticos de los materiales (CMA) y los parámetros críticos del proceso (CPP) en correlación con su efecto sobre atributos críticos de calidad (CQA) es muy recomendable para desarrollar una plataforma.
Además, las materias primas (definidas como materiales auxiliares en USP <1043>) utilizados durante la fabricación y el almacenamiento influirá significativamente en la caracterización y procesabilidad de un ATMP. Los materiales se definen como materias primas (auxiliares) si se utilizan en la proceso de fabricación de productos de ingeniería genética, celular o tisular, pero no son que se pretende que forme parte del producto final. Por ejemplo: suero fetal bovino, enzimas de digestión (por ejemplo, colagenasa, DNAse), factores de crecimiento, citoquinas, anticuerpos monoclonales, antibióticos, resinas, dispositivos y medios de separación celular componentes.1
Selección de los materiales de partida y de partida apropiados materiales es un paso crucial para evitar retrasos, repeticiones de trabajo o costes imprevistos durante la ampliación. Además, la elección puede depender de normas o requisitos establecidos por los reguladores y la industria.
¿Cómo seleccionar las materias primas adecuadas?
La selección de las materias primas debe ser basado en una selección exhaustiva del proveedor, la caracterización del material y la idoneidad probada del material para la fabricación prevista proceso. La cadena de suministro de las materias primas debe ser segura y fiable. Para garantizarlo, debe realizarse una calificación del proveedor y la calidad Hay que formular contratos de acuerdo. Documentos designados para consultar seleccionar las materias primas y poner en marcha su proyecto de desarrollo son:
- EudraLex volumen 4, parte IV capítulo 7 materias primas y de partida
- Capítulo general sobre las materias primas de origen biológico para la producción de medicamentos de base celular y de terapia génica productos (PE 5.2.12)
- USP Células y genes productos terapéuticos
- USP Terapia génica Productos
- USP Materiales auxiliares para productos de ingeniería celular, genética y tisular. Conferencia obligatoria
Además, destacamos algunos temas específicos para guiarle en el proceso de toma de decisiones:
- Fuente de materia prima
¿Su materia prima es de origen sanguíneo humano o animal, producida con sustancias de origen humano u original, o está libre de sustancias de origen humano o animal?
Es importante minimizar el riesgo de transmisión de agentes adventicios. Dado que en la producción de ATMP se utilizan materiales de partida derivados de la sangre y los tejidos humanos, el rastreo y las pruebas (de los donantes) para detectar agentes infecciosos transmisibles es un elemento clave (de acuerdo con la directiva 2004/23/CE y 2002/98/CE). Los aspectos de seguridad viral, tal y como se especifica en el texto general del PE 5.1.7, también deben ser considerados y respetados.
Se puede encontrar más información en la nota de orientación sobre la minimización del riesgo de transmisión de agentes de encefalopatía espongiforme animal a través de medicamentos humanos y veterinarios (EMA/410/01 rev.3), la declaración de posición del CHMP/CAT sobre la enfermedad de Creutzfeldt-Jakob y los medicamentos de terapia avanzada (EMA/CHMP/BWP/353632/2010) y las Directrices de la OMS sobre la distribución de la infectividad tisular en las encefalopatías espongiformes transmisibles. - Proceso de producción
Las materias primas deben tener certificados de pruebas de esterilidad o contaminación microbiana conocida, nivel de endotoxinas, impurezas y otros requisitos de calidad predefinidos.
Dado que las terapias celulares no pueden ser esterilizadas de forma definitiva por filtración de membrana, los pasos de producción se realizan en un LAF de grado A con fondo B o en un aislador con fondo C o D. En algunas condiciones, se pueden permitir otros grados de zona limpia, que se especifican en el capítulo 9.5 del volumen 4 de EudraLex, parte IV.2 Debido a esta limitación de la esterilización, se aplican estrictos requisitos de abastecimiento y aceptación. - Requisitos de calidad
Los requisitos de calidad de las materias primas deben definirse previamente en cuanto a su identidad, pureza y actividad biológica.
Por ejemplo, la osmolaridad (EP 2.2.35), el pH (EP 2.2.3), el micoplasma (EP 2.6.7), las endotoxinas (EP 2.6.14) deben medirse y analizarse para cumplir los requisitos necesarios. Los reguladores admiten que, a menudo, las materias primas sólo están disponibles con la declaración de grado de investigación2 y su calidad, seguridad y consistencia es difícil de evaluar. Los sueros de origen humano o animal son ejemplos típicos de mezclas biológicas complejas cuya composición no siempre puede determinarse o definirse completamente. Los fabricantes deben ser conscientes de estos riesgos y proporcionar detalles sobre la idoneidad y el posible uso del material. Estos detalles deben incluir/centrarse en la identidad, la seguridad y la funcionalidad del material. Sin embargo, esta información combinada suele faltar 2.
El proceso de seleccionar las materias primas adecuadas y, al mismo tiempo, tener en cuenta todas las directrices vigentes puede ser todo un reto. Los experimentados consultores de QbD pueden ayudarle a cumplir estos requisitos reglamentarios de los ATMP. Nuestro formato de célula por diseño es capaz de evaluar el impacto de la variabilidad de la materia prima, identificar los parámetros críticos del proceso y definir los atributos críticos de calidad del producto. No dude en ponerse en contacto con nosotros para obtener más información.
1EMA/CAT/852602/2018Proyecto de directriz sobre los requisitos de calidad, no clínicos y clínicos de los medicamentos en investigación de terapia avanzada en ensayos clínicos.
2ParteIV del volumen 4 de EudraLex Directrices sobre prácticas correctas de fabricación específicas para los productos avanzados
Medicamentos terapéuticos.