
Los dispositivos médicos de diagnóstico in vitro (DIV ) llevan mucho tiempo formando parte de nuestra industria sanitaria. Sin embargo, en los últimos tiempos el uso de los DIV ha aumentado drásticamente. Basta con pensar en todas las pruebas de diagnóstico del SARS-CoV2 que se han realizado en los últimos 2 años.
Teniendo en cuenta la rápida propagación del coronavirus y sus implicaciones, es fácil ver cómo los resultados falsos negativos de las pruebas pueden tener consecuencias catastróficas. Por ello, es muy importante que estas pruebas de diagnóstico sean fiables.
Para muchos otros DIV, las consecuencias de los resultados incorrectos son igualmente amenazantes. Por lo tanto, es esencial que los DIV estén regulados para garantizar la seguridad del público y de los pacientes. Sin embargo, dado que la definición formal de DIV (véase más adelante) abarca una gama tan amplia, es ilógico imponer la misma normativa a todos.
En consecuencia, se introdujo una subdivisión en cuatro clases diferentes. Este artículo demostrará a fondo cómo se clasifican los DIVs según el IVDR, ya que esta clasificación es responsabilidad de los fabricantes.
Definición de DIV y clasificación de DIV
¿Qué son los "productos sanitarios de diagnóstico in vitro" o DIV?
«Dispositivo médico de diagnóstico in vitro» significa
cualquier producto sanitario que sea un reactivo, producto reactivo, calibrador, material de control, kit, instrumento, aparato, pieza de equipo, programa informático o sistema, utilizado solo o en combinación, destinado por el fabricante a ser utilizado in vitro para el examen de muestras, incluidas las donaciones de sangre y tejidos, derivadas del cuerpo humano, con el fin exclusivo o principal de proporcionar información sobre uno o varios de los siguientes aspectos
- (a) relativa a un proceso o estado fisiológico o patológico,
- (b) sobre deficiencias físicas o mentales congénitas,
- (c) sobre la predisposición a una condición médica o una enfermedad,
- (d) determinar la seguridad y la compatibilidad con los posibles receptores,
- (e) para predecir la respuesta o las reacciones al tratamiento
- o f) para definir o controlar las medidas terapéuticas».
De IVDD a IVDR: ¿qué, cuándo y por qué?
Hasta mayo de 2017 no se sometió a los DIV a una normativa europea uniforme , IVDR 2017/746 . Esta normativa se introdujo como una ampliación de las anteriores directivas sobre DIV ( DIRECTIVA DIV 98/79/CE ), por lo que, en comparación con la DIV, no se eliminaron requisitos, sino que se introdujeron otros adicionales.
Las autoridades europeas han concedido un período de transición de cinco años para que los fabricantes adapten sus modos de funcionamiento a los nuevos requisitos del IVDR, por lo que éste entrará en vigor el 26 de mayo de 2022.
Mientras que la DIV sólo impuso objetivos que se transpusieron posteriormente a la legislación nacional, la normativa del DIV tiene directamente fuerza legal vinculante en todos los Estados miembros europeos, lo que garantiza la uniformidad en toda Europa. Sin embargo, hay que tener en cuenta que los Estados miembros aún pueden imponer más restricciones a determinados DIV además de la normativa europea.
Clasificación del IVDR basada en el riesgo
El rigor de las regulaciones del IVDR impuestas a un DIV específico depende completamente de su clase de riesgo asignada. En el marco de la DIV, sólo los DIV mencionados en dos extensas listas fueron clasificados en las dos primeras clases de riesgo.
El resto de DIVs se dividieron en la categoría de «autodiagnóstico» o en la de «otros DIVs«. Según el IVDR, ahora se clasifican en categorías que van de la A (bajo riesgo) a la D (máximo riesgo), sobre la base de una serie de normas establecidas en el anexo VIII del IVDR. Estas normas se explican con más detalle en este Documento de orientación europeo .
La figura siguiente ilustra el enfoque basado en listas en el marco de la DIV, en contraste con el procedimiento de clasificación de DIV basado en el riesgo.
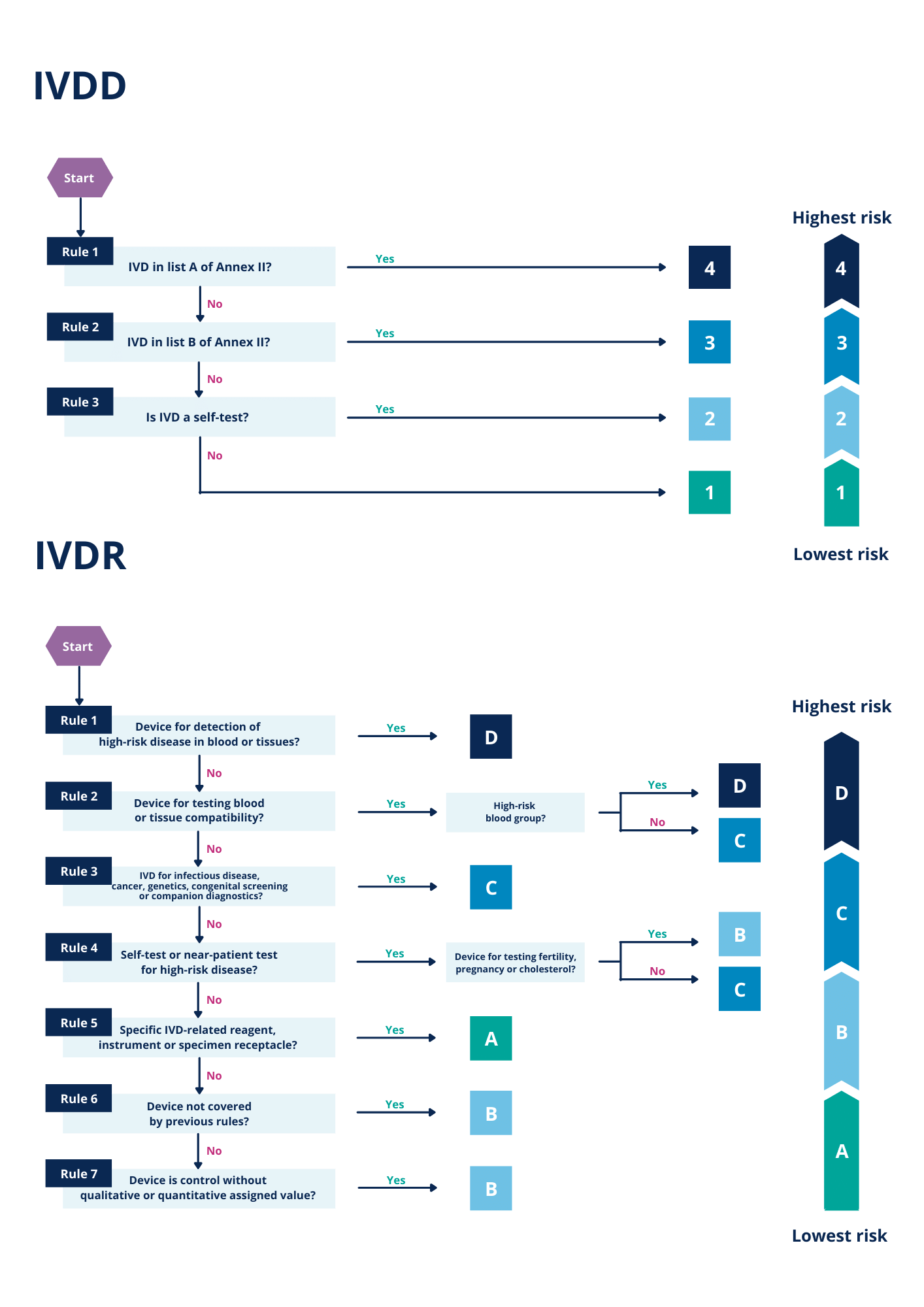
Figura 1 – Diagramas de flujo que describen el proceso de clasificación en el marco de la DIV y la DIVR.
Según el enfoque basado en la lista que utiliza la DIV, la mayoría de los DIV se incluyeron en la clase de menor riesgo («DIV otros»), que no requiere la aprobación ni la participación de un organismo notificado. El procedimiento de clasificación más extenso del IVDR categoriza una fracción significativamente mayor de todos los IVDs en clases de riesgo elevado.
Estas clases de mayor riesgo también están sujetas a la aprobación de los organismos notificados. La figura 3 ilustra este cambio de productos de clases de bajo riesgo, que no requieren aprobación, a clases de mayor riesgo que sí requieren la aprobación de un organismo notificado. Por lo tanto, para muchos DIV el rigor de los requisitos aumenta notablemente en comparación con antes de la introducción del IVDR.
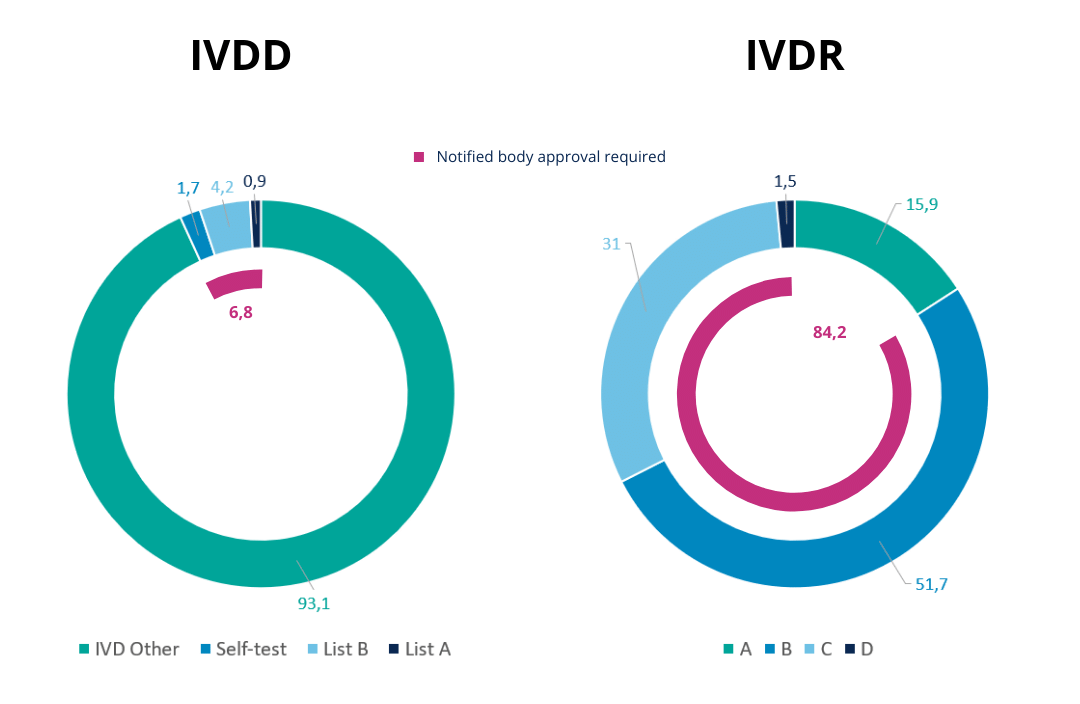
Figura 2 – Comparación entre las proporciones de clasificación en el marco de la DIVD frente a la DIVR. [Fuente]
Para poner esto en perspectiva, podemos echar un vistazo al ejemplo de las pruebas de diagnóstico del SARS-CoV2 y queda claro el efecto que puede tener esta nueva clasificación del IVDR. En el marco de la DIV, estas pruebas entrarían en la clase de menor riesgo, mientras que se clasifican en la clase D, la de mayor riesgo, bajo la IVDR . Por lo tanto, la fabricación de pruebas para el SARS-CoV2 requiere, entre otras cosas, la aprobación de un organismo notificado y que los lotes individuales sean analizados por un laboratorio de referencia.
Se acerca la fecha límite del DIV: ¿ahora qué?
El nuevo IVDR aporta uniformidad a la regulación de la amplia gama de DIVs, garantizando la seguridad de los pacientes y del público en general en toda Europa.
Gracias a la introducción de un sistema de clasificación de DIVs más completo y basado en normas, aproximadamente el 84% de los DIVs tendrán que ser aprobados por un organismo notificado, frente a menos del 7% anterior.
La aplicación del IVDR ha dado lugar a la necesidad de reclasificar, así como de certificar o recertificar, muchos DIV.
¿Aún no está seguro de cómo proceder? Nuestros expertos estarán encantados de ayudarle en la transición de la DIV a la DIV. No dude en ponerse en contacto con nosotros.