







.png?width=109&height=108&name=Pharma%20(2).png)
.png?width=111&height=108&name=Medical%20Devices%20(2).png)
.png?width=84&height=107&name=IVD%20(2).png)

.png)
Risk management is key to promoting the safety of medical devices. So it’s with good reason that the new European medical device regulations MDR and IVDR (EU regulations Regulation (EU) 2017/745 and 2017/746) emphasize this aspect. Contrary to the medical device directives’ (MDD, AIMDD, IVDD) vagueness on the matter, the MDR and IVDR address risk management requirements very specifically.
Many still believe that an FMEA ticks the risk management box.
Nevertheless, as crucial as medical device risk management may be, its implementation and subsequent application are not always straightforward. Many still see risk management as a mandatory task and believe that an FMEA (Failure Mode and Effect Analysis) ticks the box. However, this does not comply with what the regulations expect.
Medical Device Regulation (MDR)
According to the MDR (Regulation (EU) 2017/745), “risk management shall be understood as a continuous iterative process throughout the entire lifecycle of a device, requiring regular systematic updating”.
Note that this section only speaks of the requirements outlined in the MDR, but the requirements outlined in the IVDR (Annex I – General Safety and Performance Requirements – Chapter I) are equal.
The framework – in terms of risk management requirements – is outlined in the General Safety and Performance Requirements (GSPR) in Annex I of the MDR and states that manufacturers shall:
- establish and document a risk management plan for each device;
- identify and analyze the known and foreseeable hazards associated with each device;
- estimate and evaluate the risks associated with, and occurring during, the intended use and during reasonably foreseeable misuse;
- eliminate or control these risks
- evaluate the impact of information from the production phase and, in particular, from the post-market surveillance system, on hazards and the frequency of occurrence thereof, on estimates of their associated risks, as well as on the overall risk, the benefit-risk ratio, and risk acceptability; and
- based on the evaluation of the impact of this information, amend control measures if necessary.
There’s more to compliance than FMEA
An FMEA is probably the most widespread risk management tool within the MD field. Having only an FMEA (or even multiple FMEAs) in place does not make you compliant with the regulations outlined.
Why? For example, an FMEA looks at risks related to failures, while you can also expect to identify and analyze hazards during the normal intended use of the medical device. Risks associated with a medical device are not exclusively the result of failures. A device may still put patients at risk while functioning normally.
You’re expected to start with risk management at the start of the design and development process.
In addition, during an FMEA you will for example analyze the device’s components (design FMEA) or its manufacturing process (process FMEA). Therefore, your design or process must already be quite mature. And, as a result, this analysis is done in a later stage of your development trajectory.
However, you’re expected to start with risk management at the start of the design and development process. Consequently, the results or risk control measures can already be implemented during the design and development trajectory. These shall be used as design inputs.
FMEAs are very powerful and useful tools, but keep in mind that they are a component of a larger risk management system. What does such a system look like and how can you implement it in your company? The ISO 14971 standard (application of risk management to medical devices) provides you with an answer.
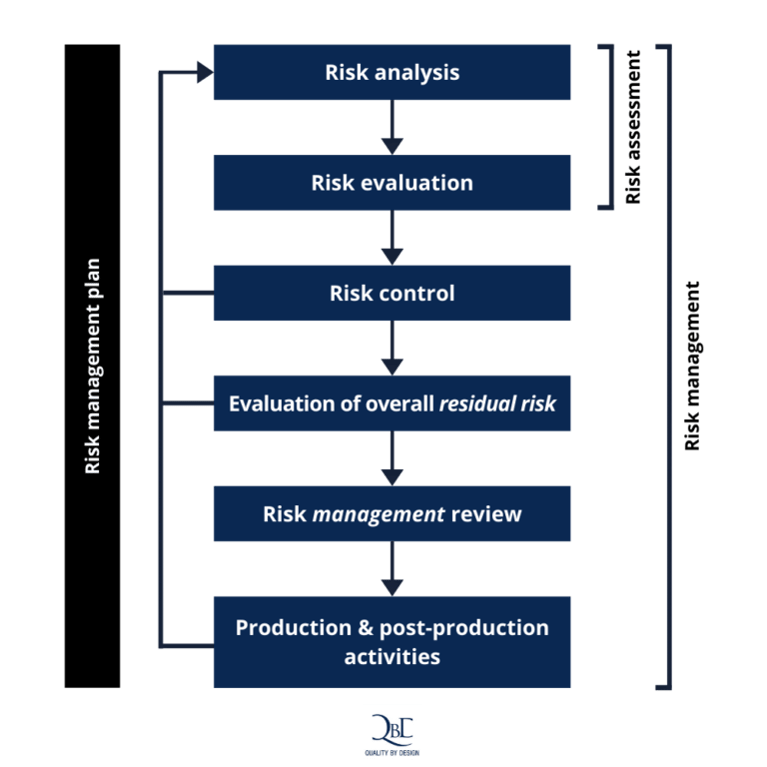
Figure 1 – A schematic representation of the risk management process
Risk management standard ISO 14971:2019
ISO 14971 is the global standard for medical device risk management. It provides a framework in which experience, insight and judgment are applied systematically to manage the risks associated with the use of medical devices. The requirements outlined in this standard are applicable to all phases of the lifecycle of a medical device.
The most recent version was published in December 2019, ISO14971:2019. Six months later, related guidance document ISO/TR 24971:2020-06 followed. This guide can be seen as a long commentary, as it concretizes the requirements of ISO 14971 and provides assistance for manufacturers of medical devices in the implementation.
The first thirty pages comment on ISO 14971:2019 chapter by chapter. This is followed by 8 appendices of 55 pages. The table below shows the contents of both documents.
ISO 14971:2019
Scope
- Normative references
- Terms and definitions
- General requirements for risk management system
- Risk analysis
- Risk evaluation
- Risk control
- Evaluation of overall residual risk
- Risk management review
- Production and post production activities
Plus:
- Annex A – Rationale for requirements
- Annex B – Risk management process for medical devices
- Annex C – Fundamental risk concepts
ISO/TR 24971:2020
Sections 1-10 correlate with ISO14971:2019
Plus:
- Annex A – Identification of hazards and characteristics related to safety
- Annex B – Techniques that support risk analysis
- Annex C – Relation between the policy, criteria for risk acceptability, risk control, and risk evaluation
- Annex D – Information for safety and information on residual risk
- Annex E – Role of international standards on risk management
- Annex F – Guidance on risks related to security
- Annex G – Components and devices designed without using ISO 14971
- Annex H – Guidance on in vitro diagnostic medical devices
ISO 14971:2019 represents the current state-of-the-art for risk management.
The ISO 14971:2019 standard was not harmonized when this blog was published. Therefore, compliance with this standard is currently not mandatory for CE marking medical devices under the European Medical Device Regulations (IVDR and MDR).
Nevertheless, ISO 14971:2019 represents the current state-of-the-art for risk management. Therefore, its application – from the start of the device development process onwards – is considered key when implementing a risk management process in an MD company.
Throughout the entire lifecycle
The medical devices regulations (MDR and IVDR) impose a continuous iterative risk management process throughout the entire lifecycle of a device. ISO 14971:2019 and the accompanying guidance document lay out such a risk management framework.

