







.png?width=109&height=108&name=Pharma%20(2).png)
.png?width=111&height=108&name=Medical%20Devices%20(2).png)
.png?width=84&height=107&name=IVD%20(2).png)

For Medicinal Products, the Product Information (PI) is a key document that provides a summary of officially approved information for Healthcare Professionals (HCPs) and Patients. The precision and clarity of the document is crucial, making the accurate and appropriate translations of the PI for medicinal products essential.
This is where the European Medicines Agency’s or EMA’s Linguistic Review comes into play — a crucial process ensuring that every word in the documentation provided to patients and healthcare providers across Europe is accurate, consistent and compliant with regulatory requirements.
In this blog post, we provide some key background on the PI for medicinal products and explain the EMA’s linguistic review process and timelines in more detail.
What is the Product Information (PI)?
For products approved via the EU Centralised Procedure (CP), the Product Information (PI) is a key document that provides a summary of officially approved information for healthcare professionals (HCPs) and patients regarding a medicine.
The PI comprises the following key documents:
- The Summary of Product Characteristics (SmPC), which forms the basis of information for HCPs on how to use the medicine safely and effectively within the terms of the marketing authorisation (MA).
- For products approved via the Centralised Procedure, Annex II contains information on any specific conditions or restrictions for use.
- Labelling Text – used to prepare the immediate or outer packaging of the medicinal product, such as blister foil, bottle label, or carton.
- The Package Leaflet, which contains information aligned with the SmPC but is presented in lay terms suitable for end-users, such as patients or carers, to read and interpret. A printed copy is enclosed with the medicine as supplied for sale.
Final versions of the PI are submitted to the European Medicines Agency (EMA) as a single document containing all of the above sections in PDF format, with specific formatting conventions required.
What is the EMA’s Linguistic Review Process?
The EMA’s Linguistic Review is the translation and review process for all new and updated Product Information (PI) required post-opinion for all medicinal products authorized by the European Union (EU) Centralized Procedure (CP).
The EMA’s Linguistic Review procedure is a standardized 25-day process that occurs during the initial Marketing Authorization Application (MAA) process, as well as following certain post-authorization procedures affecting the PI, such as variation applications or assessment of periodic safety update reports (PSURs).
After the CHMP Opinion is issued, the English Product Information (EN PI) is translated into the 25 official EU/EEA languages. These translations are subsequently reviewed by individual Member States. This process is known as the “Linguistic Review of Product Information.”
The translations must be submitted and reviewed according to a set timetable that starts immediately after the CHMP Opinion.
- For products granted an initial Marketing Authorization (MA), the date of the CHMP Opinion is taken as Day 210, which corresponds to the end of the Marketing Authorization Application (MAA) assessment.
- For post-approval changes, Day 0 is used to define the date of the CHMP Opinion.
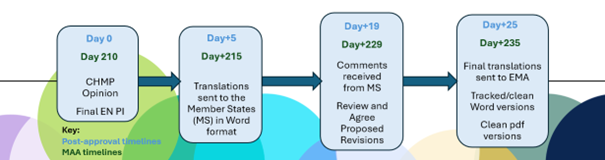
Figure 1. Linguistic Review Timelines and documentation requirements
Key Timelines in the EMA’s Linguistic Review Process:
- Day +5 / Day +215 Submission: For most procedures, once the CHMP Opinion is adopted, the applicant must submit the translations of the PI just 5 calendar days later. With the CHMP meeting concluding on a Thursday each month, there are only 3 working days to finalize the submission—a pragmatic approach to starting translations is required.
- Day +25 / Day +235 Submission: There are only 4 working days to review and finalize the submission after the deadline for Member State (MS) comments at Day +19 / Day 229. The MS comments may require dialogue between the Marketing Authorization Holder (MAH) and MS to agree on the final wording.
Preparation, Review & Approval of the PI
During an initial application for marketing authorisation, the EN PI is prepared by the applicant in accordance with Quality Review of Documents (QRD) templates and stylistic requirements.
Throughout the assessment process, the document undergoes multiple rounds of revision by the regulatory agency. All aspects of the CTD dossier are summarised in the document, including:
- Safety and efficacy information from non-clinical and clinical studies, such as the indication, posology, side effects, and information on specific populations including paediatrics and the elderly.
- Details of quality aspects such as the strength, pharmaceutical form, excipients, and the manufacturer responsible for batch release.
- Administrative information such as the Product Name, MAH details, MA numbers, and pack sizes.
For a centralised product, the EMA Committee for Human Medicinal Products (CHMP) will, following the assessment of the MAA, recommend whether the application is considered approvable.
- The positive (or negative) opinion is sent to the European Commission (EC), which ultimately grants the EU MA, typically around 67 days after the CHMP’s recommendation.
- The same principles apply to products that already have an MA — post-authorisation changes are reviewed by the EMA, with the CHMP adopting a recommendation as to whether the change is approvable.
- Again, the positive (or negative) opinion is sent to the EC, which updates the terms of the EU MA, typically around 67 days after the CHMP’s recommendation or on an annual basis, depending on the type of change applied.
QbD’s Expertise in Streamlining Linguistic Review Processes
Clients are often rightly focused on achieving the principal aim of MAA or variation approval and may not give sufficient consideration to the very time- and resource-intensive process of post-approval Linguistic Review of Product Information.
It is important to note that the PI will be the text used to prepare the artwork and labelling for commercial supply; therefore, ensuring a high standard of accuracy and compliance across all languages is imperative.
QbD’s flexible end-to-end service offering can provide support in the preparation, review, and submission of the PI translations, managing MS review comments until the final agreed translations are submitted at the end of the process.
The QbD Group has a wealth of experience in this area. Over the past decade, we have project-managed more than 250 Linguistic Reviews. Our clients trust us to manage the entire linguistic review process, and we excel at the following activities:
- Reviewing the EN PI to ensure that the content complies with QRD stylistic and formatting guidelines prior to the initial MAA or post-approval submission.
- Managing project aspects, including determining when to initiate work on the initial translations to meet the submission deadline. As highlighted earlier, at the time of CHMP Opinion, there are only three working days before submission for MS review.
- Coordinating initial translations, implementing effective quality control steps, delivering files to clients for local review, and submitting them for MS review in accordance with strict procedural timelines.
- Utilizing well-defined standard processes and software applications to ensure accurate content and formatting, all of which ensures that we meet submission deadlines.
- Drawing on our extensive knowledge and experience with the requirements for various EMA Procedures, including managing updates across product families, worksharing, and implementing changes for Centrally-Authorised and Nationally-Authorised Products following PSUSA or Referral procedures.
Summary and Future Considerations
The preparation of high-quality English Product Information (PI) is a crucial starting point for developing translations as part of the time-sensitive and resource-intensive Linguistic Review process. It is important to note that the PI is subsequently used to prepare artwork and labelling for commercial production.
Looking ahead, the development of electronic Product Information (ePI) is likely to impact the current process described in this article. In a future post, we will explore the potential impact of ePI on Regulatory Agencies, Marketing Authorization Holders (MAHs), Healthcare Professionals (HCPs), and patients.


%20Checklist.jpg)







.jpg)




.jpg)
.jpg)

.jpg)


.jpg)
.jpg)
.png)