

In vitro diagnostic medical devices (IVDs) have long been a part of our healthcare industry. Recently, however, the use of IVDs has increased drastically. Just think of all SARS-CoV2 diagnostic tests that have been performed over the last 2 years.
Keeping in mind the rapid spread of the coronavirus and its implications, it is easy to see how false-negative test results can have catastrophic consequences. As a result, it is of great importance that these diagnostic tests are reliable.
For many other IVDs, the consequences of incorrect results are equally threatening. Therefore, it is essential that IVDs are regulated in order to ensure public and patient safety. However, since the formal definition of IVDs (see below) spans such a broad range, it is illogical to impose the same regulations on all.
Consequently, a subdivision into four different classes was introduced. This article will thoroughly demonstrate how IVDs are classified according to IVDR, since this classification is the manufacturers’ responsibility.
| ## IVD definition and IVDR classification<br>### What are ‘in vitro diagnostic medical devices’ or IVDs?<br>“ In-vitro diagnostic medical device” means<br>‘any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information on one or more of the following:<br>- (a) concerning a physiological or pathological process or state,<br>- (b) concerning congenital physical or mental impairments,<br>- (c) concerning the predisposition to a medical condition or a disease,<br>- (d) to determine the safety and compatibility with potential recipients,<br>- (e) to predict treatment response or reactions<br>- or (f) to define or monitor therapeutic measures.’ |
IVDD to IVDR: what, when, and why?
It was only in May 2017 that IVDs were first subjected to uniform European regulations, IVDR 2017/746. These regulations were introduced as an extension of the prior IVD directives (IVDD 98/79/EC), so compared to IVDD no requirements were removed, but additional ones were introduced.
European officials allowed a five-year transition period for manufacturers to adapt their modes of operation to the new IVDR requirements, hence, the IVDR will come into effect on May 26th of 2022.
While IVDD only imposed objectives that were subsequently transposed into national legislation, the IVDR regulations directly have binding legal force in all European member states, thus ensuring uniformity across Europe. Be aware, however, that member states can still impose further restrictions to certain IVDs on top of the European regulations.
Risk-based IVDR classification
The strictness of the IVDR regulations imposed on a specific IVD is completely dependent on its assigned risk-class. Under the IVDD, only IVDs mentioned in two extensive lists were classified in the top two risk classes.
The remaining IVDs were then divided into either the ‘self-test’ category or ‘IVD others’ category. Under IVDR they are now classified in categories from A (low risk) to D (highest risk), based on a set of rules stated in Annex VIII of the IVDR. These rules are further explained in this European guidance document.
The figure below illustrates the list-based approach under IVDD in contrast to the risk-based IVDR classification procedure of IVDs.
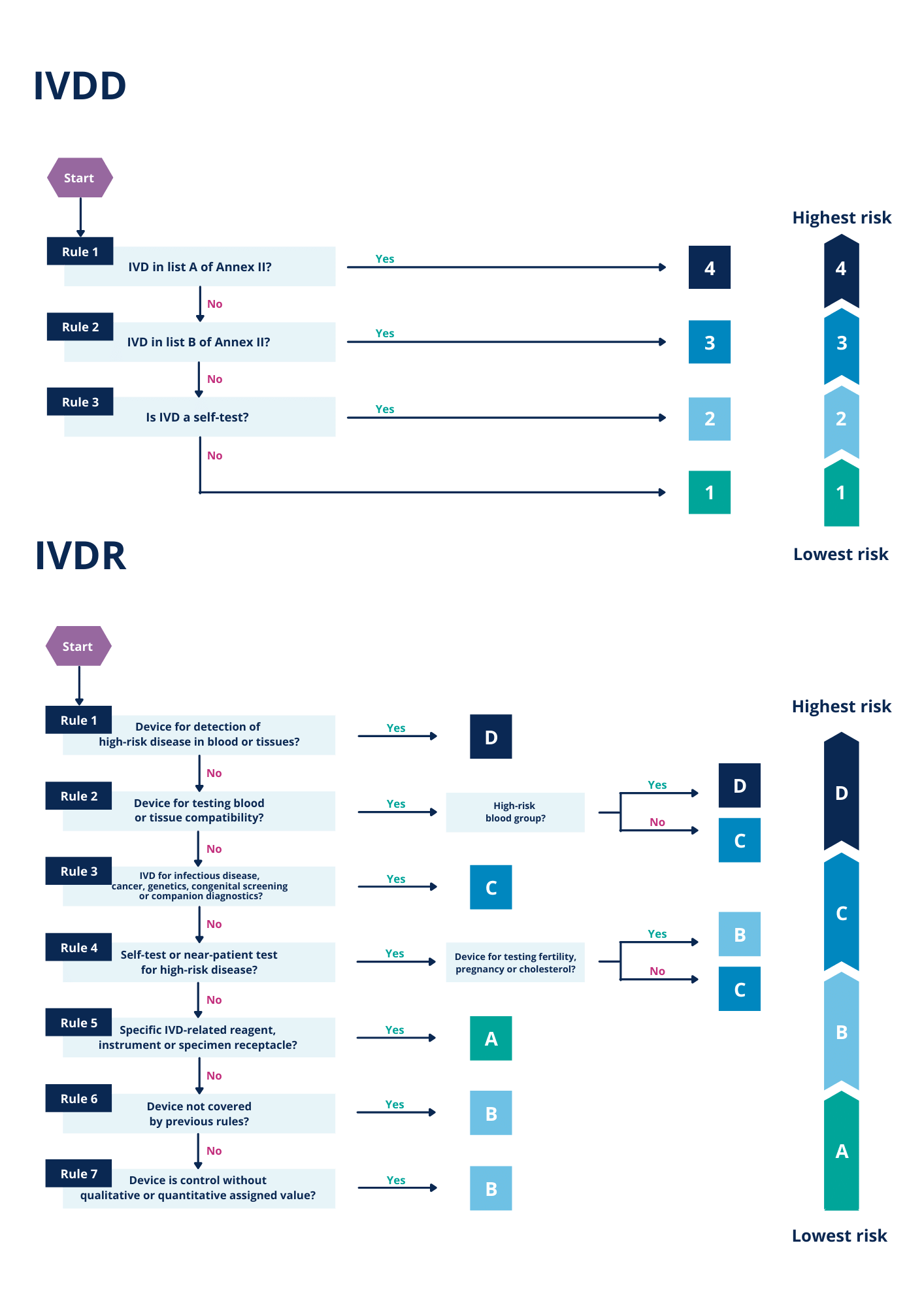
Figure 1 – Flowcharts describing the classification process under IVDD and IVDR.
According to the list-based approach used by the IVDD, most of the IVDs were put into the lowest risk class (‘IVD others’) which does not require the approval or involvement of a notified body. The more extensive IVDR classification procedure categorizes a significantly larger fraction of all IVDs in elevated risk classes.
These higher-risk classes are also subject to the approval of notified bodies. Figure 3 illustrates this shift of products from classes of low risk, not requiring approval, to classes of higher risk which do require approval of a notified body. Hence, for many IVDs the stringency of the requirements increases notably compared to before the introduction of the IVDR.
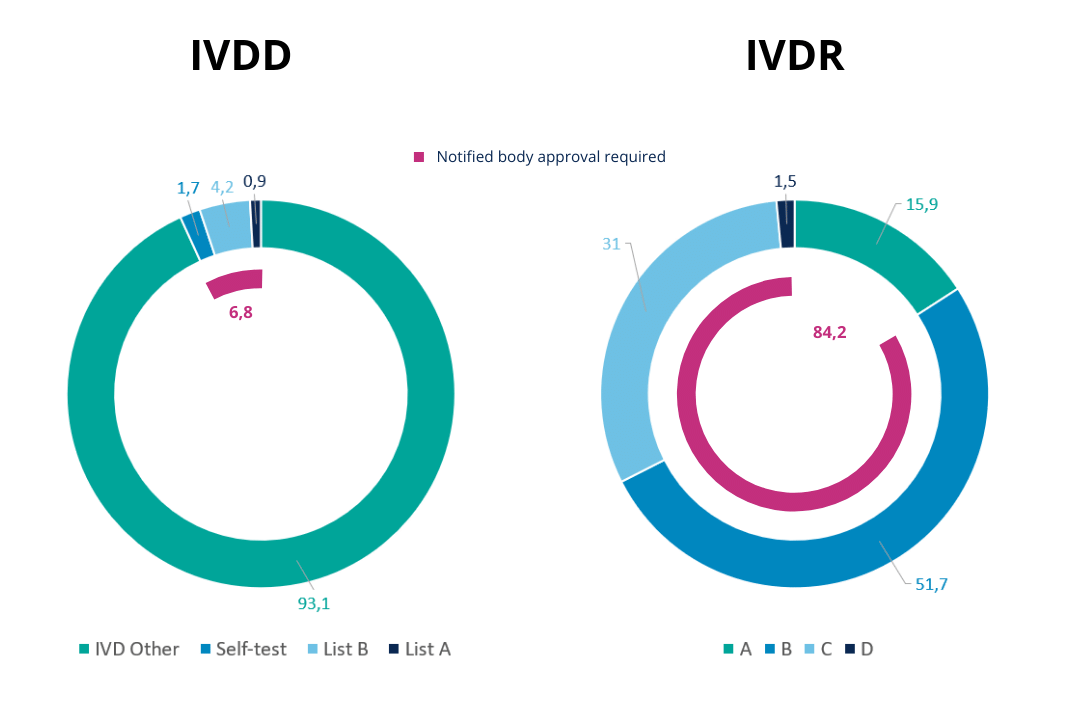
Figure 2 – Comparison between classification proportions under IVDD vs IVDR. [ Source]
To put this into perspective, we can take a look at the example of SARS-CoV2 diagnostic tests and it becomes clear how much effect this new IVDR classification can have. Under IVDD, these tests would fall under the lowest risk class, while they are classified in class D, the highest risk class, under IVDR. Consequently, the manufacturing of SARS-CoV2 tests requires among others notified body approval and individual batches to be tested by a reference laboratory.
IVDR deadline approaching: what now?
The new IVDR brings uniformity to the regulation of the broad range of IVDs, ensuring the safety of patients and the general public across all of Europe.
Thanks to the introduction of a more complete rule-based IVDR classification system approximately 84% of IVDs will now need to be approved by a notified body, compared to less than 7% before.
The implementation of the IVDR has resulted in the need for reclassification as well as certification or recertification for many IVDs.
Still unsure on how to proceed? Our experts are happy to assist you in the transition from IVDD to IVDR. Do not hesitate tocontact us .
About the Author

About the Author

Life Science Consultant at QbD Group
Ward is a Life Science Consultant at QbD Group, providing expert guidance on quality, regulatory, and compliance challenges in the pharmaceutical industry.

Navigate Regulatory Complexity with Confidence
Navigate complex regulatory landscapes with expert guidance across pharmaceuticals, medical devices, and IVD.
Read moreSubscribe to the latest updates in life science
Expert perspectives delivered to your inbox — pick your interests.
No spam, ever. Unsubscribe anytime.


