







.png?width=109&height=108&name=Pharma%20(2).png)
.png?width=111&height=108&name=Medical%20Devices%20(2).png)
.png?width=84&height=107&name=IVD%20(2).png)

The first step: the responsibilities of the Manufacturer
The manufacturer must establish a quality management system in accordance with the MDR’s requirements. This system should be documented, implemented, and effective throughout the life cycle of the medical device.
The Notified Body will then request all necessary information and data to conduct an audit of the quality management system, ensuring it meets the specified requirements. The following aspects must be documented:
- Manufacturer’s quality objectives,
- Organization of the company,
- Procedures and techniques for monitoring, verification, validation, and control of product design,
- Quality assurance and control techniques for manufacturing, and
- Testing procedures before, during, and after manufacturing.
Furthermore, the manufacturer must also provide the technical documentation. If other standards are applied by the manufacturer, conformity with these standards will also be assessed. If the audit is successfully passed, an EU Quality Management Certificate is issued by the Notified Body.
The next step: the Responsibilities of the Notified Body
A surveillance assessment is carried out for Class IIa, IIb, and III products (including Im, Is, Ir categories). The Notified Body will perform regular audits and assessments (usually annually, as well as unannounced at least once every 5 years) to ensure that the manufacturer continues to apply the previously approved Quality Management (QM) system and additionally implements the post-market surveillance plan. For Class IIa and IIb devices, the surveillance assessment also includes an evaluation of the technical documentation.
The certificate of conformity issued by the Notified Bodies is valid for a maximum of 5 years. This certificate can be extended upon the manufacturer’s application, accompanied by a reassessment. Under MDR, the CE mark Certificates are required to be registered in EUDAMED (the European Electronic Database on Medical Devices).
What about clinical evaluation and the different classes of devices?
The proof of compliance with the General Safety and Performance Requirements for each medical device is under increased scrutiny. Since the implementation of the MDR (Medical Device Regulation), a more detailed and structured process has been mandated, dictating the need for sufficient clinical evidence for all medical devices, regardless of their classification.
Clinical Evaluation is an integral part of the Technical Documentation. The general requirements for the clinical evaluation process are outlined in paragraph 3 of Article 61. This process is a defined and methodologically sound procedure that must include:
- A critical evaluation of the relevant scientific literature,
- A critical evaluation of the results of all available clinical investigations, and
- Consideration of currently available alternative treatment options.
As per paragraph 1 of Part A of Annex XIV, there should be a Clinical Evaluation Plan, implying a planning stage. This includes a stage of identifying relevant clinical data (Stage 1), a stage of appraising the data (Stage 2), and, depending on the appraisal’s outcome, potentially generating new data through clinical investigations. A stage of analyzing the data (Stage 3) follows. Finally, as per paragraph 4, the process and outcome must be documented in a Clinical Evaluation Report, signifying a reporting stage (Stage 4).
This process can follow the guidelines of MEDDEV 2.7/1 rev4 until specific guidance on Clinical Evaluations is issued by the MDCG (Medical Device Coordination Group). Note that no exemptions are made based on the device’s classification, indicating that all types of medical devices are required to undergo the same process for clinical evaluation. This involves a state-of-the-art literature review and the formulation of a clinical evaluation plan and report.
Need assistance with CE approval for your Medical Device?
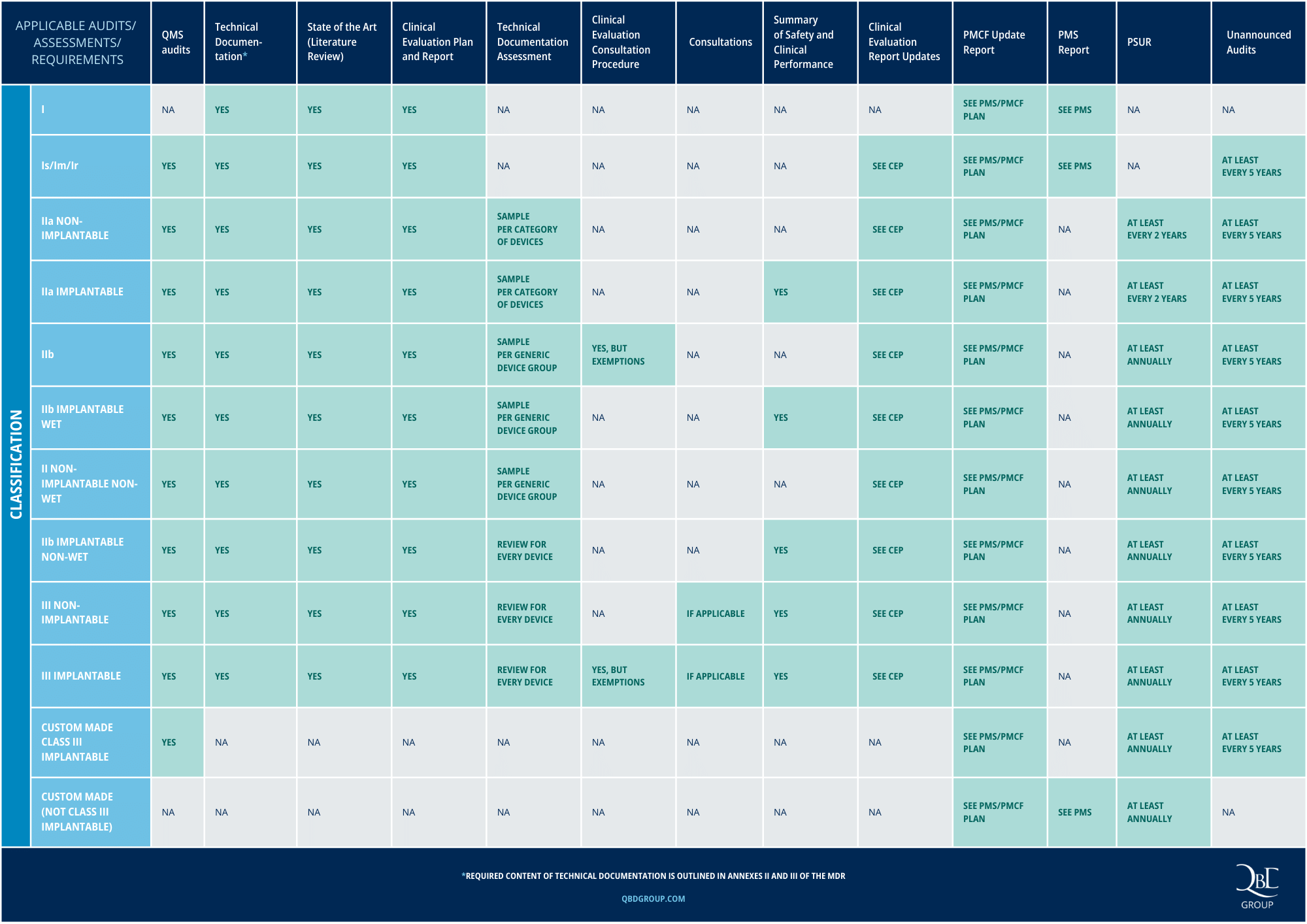
The conformity assessment procedure, with its various variants and requirements under MDR, can seem daunting. In the table below (click to enlarge), we have summarized all requirements in a concise manner, categorized by device classification.









.jpg)



