.png?width=109&height=108&name=Pharma%20(2).png)
.png?width=111&height=108&name=Medical%20Devices%20(2).png)
.png?width=84&height=107&name=IVD%20(2).png)




.png)
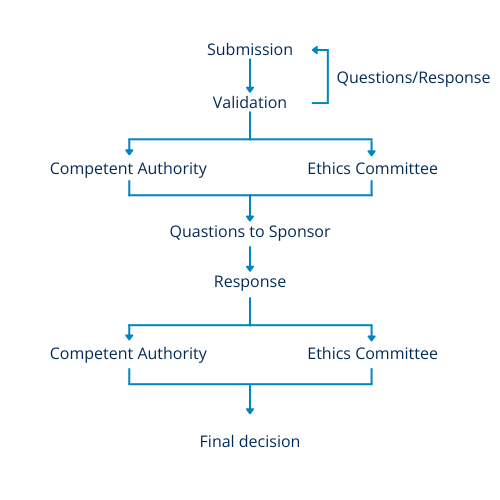
In both systems, a single set of questions is received, and a single set of responses is submitted. Figure 1 offers a summary of the two processes utilized in both the UK and EU for reviewing clinical trial applications. The first stage is an initial validation check to ensure that all necessary documents are included. Following this, the documents are made accessible to the appropriate regulatory bodies for review.
Questions that arise during the review process are gathered and presented as a single set to the sponsor. However, in the UK, attending the meeting with the Ethics Committee (EC) may provide insight into the potential questions. The sponsor then provides responses to all questions through the online portal, and the information is distributed for final decision-making.
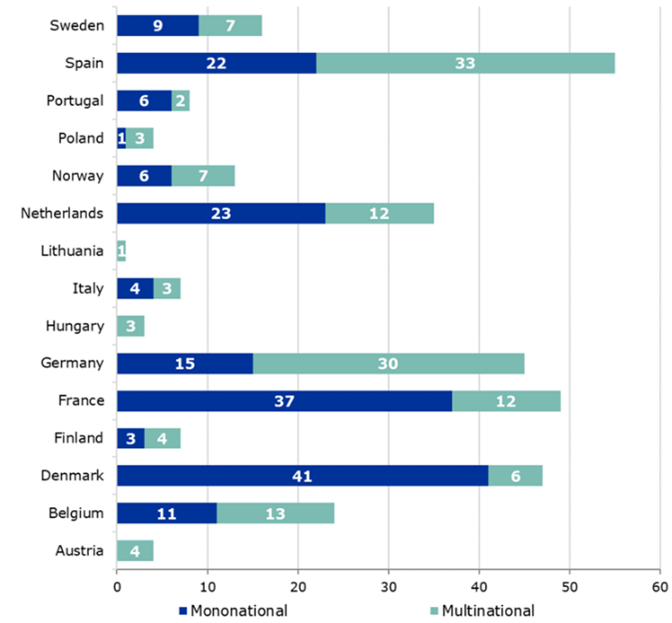
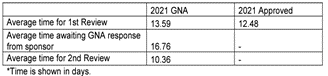
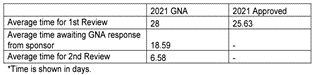
The differences between the two systems arise when looking at the timelines of the CTA process. It is easily explained by the fact that the EU systems must accommodate submissions to multiple countries. The timelines shown below represent the maximum allowable time and can be shorter on a case-by-case basis.